Decoding Enzyme Function: The Ultimate Guide to the EC Number Classification System for Biomedical Researchers
This comprehensive guide demystifies the Enzyme Commission (EC) number hierarchical classification system for researchers, scientists, and drug development professionals.
Decoding Enzyme Function: The Ultimate Guide to the EC Number Classification System for Biomedical Researchers
Abstract
This comprehensive guide demystifies the Enzyme Commission (EC) number hierarchical classification system for researchers, scientists, and drug development professionals. The article provides a foundational explanation of the EC system's four-tiered structure, explores its critical applications in modern bioinformatics and database navigation, addresses common challenges in enzyme annotation and classification, and evaluates its strengths, limitations, and modern alternatives. The content synthesizes current best practices for leveraging this essential nomenclature to drive discovery in enzymology, metabolic engineering, and drug target identification.
What Are EC Numbers? Understanding the Universal Language of Enzymes
Within the framework of a comprehensive thesis on the Enzyme Commission (EC) number hierarchical classification system, understanding its origin is paramount. The International Union of Biochemistry and Molecular Biology (IUBMB) established this standardized nomenclature to address the profound confusion that plagued enzymology in its early decades. Prior to its adoption, enzymes were named haphazardly by discoverers, leading to multiple names for the same enzyme or identical names for different enzymes. This inconsistency presented a significant barrier to scientific communication, database organization, and the burgeoning field of drug development. This whitepaper delves into the technical necessity and the enduring purpose of the EC system, providing a foundational guide for researchers and industry professionals.
Historical Imperative and Quantitative Justification
The pre-EC nomenclature landscape was characterized by redundancy and ambiguity. The following table quantifies the core issues that the IUBMB sought to resolve, based on historical analysis and contemporary reviews of the literature.
Table 1: Catalytic for Standardization: Problems in Pre-EC Nomenclature
| Problem Category | Quantitative/Qualitative Impact | Example (Pre-1961) |
|---|---|---|
| Multiple Names for One Enzyme | High frequency; one enzyme known by 3+ names in literature. | Alcohol dehydrogenase also called Alcohol:NAD+ oxidoreductase, Yeast fermenting enzyme. |
| Same Name for Different Enzymes | Led to misidentification and experimental replication failures. | Catalase referred to both peroxidase and true catalase activities. |
| Names Implying Incorrect Function | Obscured true biochemical reaction, hindering metabolic mapping. | Malic enzyme (EC 1.1.1.40) does not simply hydrolyze malate but decarboxylates it. |
| Exponential Growth of Literature | Published papers on enzymes doubled ~every 10 years (1950-1960), exacerbating naming chaos. | Necessitated a scalable, logical indexing system for information retrieval. |
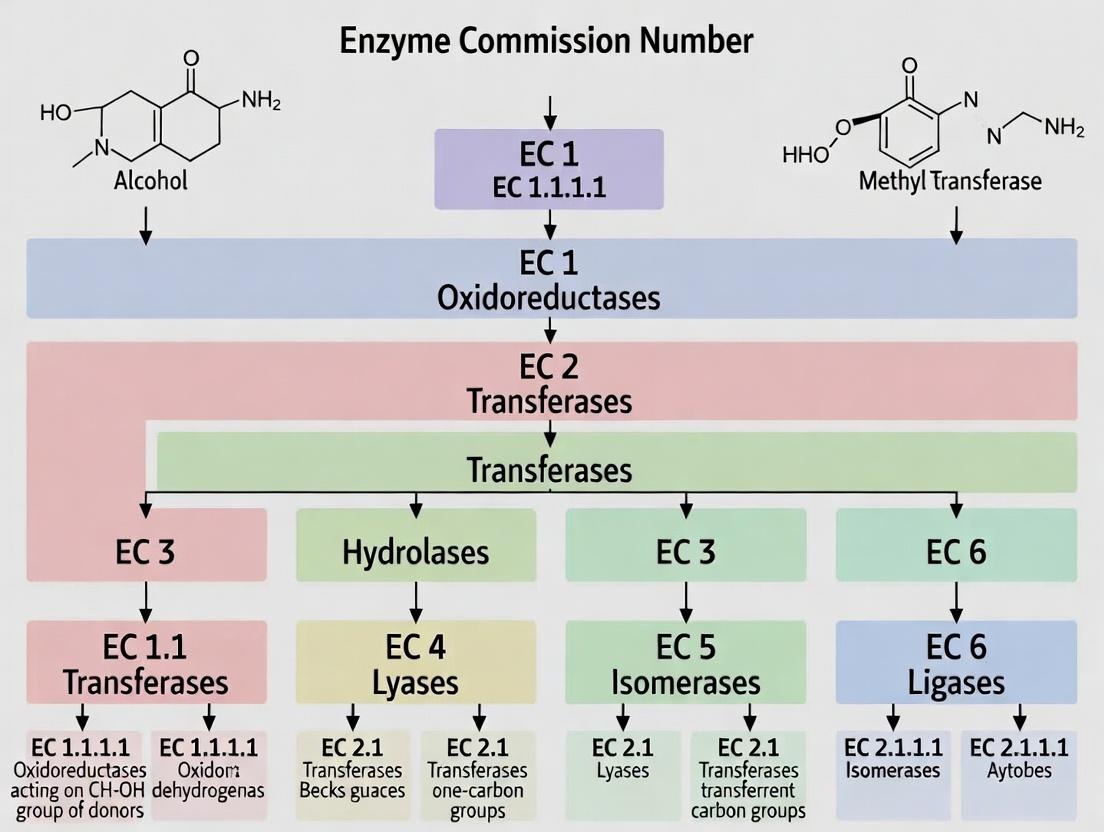
The Hierarchical Solution: EC Number Structure and Logic
The IUBMB, through its Enzyme Commission, created a four-tiered numerical classification (EC a.b.c.d) where each level provides specific, unambiguous information about the catalyzed reaction.
Table 2: The EC Number Hierarchical Framework
| EC Level | Name | Basis of Classification | Example: EC 1.1.1.1 |
|---|---|---|---|
| First Digit (a) | Class | General type of reaction (broadest category). | 1: Oxidoreductase |
| Second Digit (b) | Subclass | Specific type of donor/group involved in the reaction. | 1.1: Acting on the CH-OH group of donors |
| Third Digit (c) | Sub-subclass | Further specificity of acceptor or type of reaction. | 1.1.1: With NAD+ or NADP+ as acceptor |
| Fourth Digit (d) | Serial Number | Unique identifier for the enzyme within its sub-subclass. | 1.1.1.1: Alcohol dehydrogenase |
Experimental Protocol: Determining an EC Number for a Novel Enzyme
For researchers characterizing a new enzyme activity, the following methodology is essential for eventual EC number assignment via the IUBMB Nomenclature Committee.
Protocol: Kinetic and Specificity Profiling for EC Classification
- Purification: Homogenize source tissue/cells and purify the enzyme to homogeneity using column chromatography (e.g., affinity, ion-exchange, size-exclusion). Confirm purity via SDS-PAGE.
- Reaction Characterization:
- Determine the complete stoichiometric equation of the catalyzed reaction using HPLC or mass spectrometry to identify all substrates and products.
- Measure initial reaction rates under varied conditions (pH, temperature) to establish optimal activity.
- Class Determination (First Digit):
- Perform spectrophotometric or electrochemical assays to identify if the reaction involves oxidation-reduction (Class 1), group transfer (Class 2), hydrolysis (Class 3), etc.
- Subclass/Sub-subclass Determination (Second & Third Digits):
- Donor/Acceptor Specificity: Systematically test a panel of potential donor and acceptor molecules (e.g., different CH-OH donors, nucleotide cofactors) in coupled enzyme assays. Example: For a suspected oxidoreductase, test NAD+, NADP+, FAD, FMN, cytochrome c as electron acceptors.
- Stereospecificity: Determine if the enzyme acts on a specific stereoisomer using chiral substrates or analysis of product chirality.
- Data Submission: Compile kinetic data (Km, Vmax, kcat), substrate specificity profiles, and sequence/structure data (if available). Submit a formal recommendation to the IUBMB Enzyme Nomenclature database via the designated portal for review and assignment of a unique serial number (fourth digit).
Logical Workflow of EC Number Assignment
The following diagram illustrates the decision-making logic for classifying an enzyme, a cornerstone concept in EC system research.
Title: Logical Decision Tree for EC Class Determination
The Scientist's Toolkit: Essential Reagents for Enzyme Characterization
Table 3: Key Research Reagent Solutions for EC Classification Studies
| Reagent/Material | Function in EC Characterization |
|---|---|
| Purified Enzyme Sample | The target protein, purified to homogeneity for unambiguous activity assignment. |
| Substrate Library | A panel of chemically related compounds to test donor/acceptor specificity and determine subclass. |
| Cofactor Panel (NAD+, NADP+, ATP, etc.) | Essential for identifying the reaction mechanism and cofactor dependence (critical for Classes 1, 2, 6). |
| Coupled Enzyme Assay Systems | Enzymes like lactate dehydrogenase or pyruvate kinase, used to link the target enzyme's reaction to a measurable signal (e.g., NADH oxidation). |
| Spectrophotometer/Fluorometer | For real-time kinetic measurement of product formation or cofactor conversion (e.g., NADH at 340 nm). |
| Chiral Chromatography Columns | To determine stereospecificity of the enzyme, a key differentiator at the sub-subclass level. |
| Reference Databases (BRENDA, KEGG) | To compare kinetic parameters and substrate profiles against known, classified enzymes. |
Visualization of the EC System's Integration in Modern Research
The EC number serves as a universal key linking disparate types of biological data, a foundational principle for systems biology and drug discovery.
Title: EC Number as a Central Hub for Biological Data Integration
The IUBMB's creation of the Enzyme Commission number system was a direct, necessary response to the untenable heterogeneity of early biochemical nomenclature. By imposing a rigorous, reaction-based hierarchical logic, it provided a stable, scalable, and unambiguous framework. This standardization is not merely archival; it is the critical infrastructure that enables the computational integration of genomic, structural, kinetic, and pathway data. For the modern researcher and drug developer, the EC number remains an indispensable tool for precisely targeting enzymes, interpreting high-throughput data, and rationally designing inhibitors or biocatalysts, thereby fulfilling its original purpose as the universal language of enzymology.
The Enzyme Commission (EC) number hierarchical classification system is a formal, numerical taxonomy for enzymes, developed and maintained by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB). It is a cornerstone of systematic research in biochemistry, molecular biology, and drug development, providing a precise, machine-readable language for enzyme function. This whitepaper provides a deep technical dive into the structure and application of this four-level system, framed within ongoing research to map the catalytic landscape of life and its pharmacological modulation.
The Four-Level Hierarchical Structure
Each EC number is of the form EC X.X.X.X, where each component represents a successively more specific classification. The system operates on the principle of chemical reaction specificity.
Table 1: The Four-Tiered EC Number Hierarchy
| EC Level | Name | Description | Example (EC 1.1.1.1) |
|---|---|---|---|
| First (X.-.-.-) | Class | Broadest category, defines the type of chemical reaction catalyzed. | 1: Oxidoreductases – Catalyze oxidation/reduction reactions. |
| Second (X.X.-.-) | Subclass | Specifies the group of the donor in oxidoreductases, or the type of bond acted upon in other classes. | 1.1: Acting on the CH-OH group of donors. |
| Third (X.X.X.-) | Sub-subclass | Further specifies the type of acceptor involved. | 1.1.1: With NAD⁺ or NADP⁺ as acceptor. |
| Fourth (X.X.X.X) | Serial Number | A unique identifier for the specific enzyme/substrate combination within the sub-subclass. | 1.1.1.1: Alcohol dehydrogenase. |
The seven main enzyme classes are: 1. Oxidoreductases, 2. Transferases, 3. Hydrolases, 4. Lyases, 5. Isomerases, 6. Ligases (Synthetases), and 7. Translocases (added more recently).
Experimental Protocol: Determining an Unknown Enzyme's EC Number
A systematic approach is required to classify a novel enzyme. The following protocol outlines key methodologies.
1. Reaction Characterization and Substrate Specificity Assay
- Objective: Determine the exact chemical transformation and identify permissible substrates.
- Methodology:
- Purify the enzyme to homogeneity using chromatographic techniques (e.g., affinity, size-exclusion).
- Incubate the purified enzyme with a panel of potential substrate candidates under optimized pH and temperature.
- Use techniques like HPLC, mass spectrometry, or spectrophotometry to detect product formation for each candidate.
- Perform Michaelis-Menten kinetics (see below) to determine kinetic parameters (Km, kcat) for each viable substrate.
2. Kinetic Analysis (Michaelis-Menten)
- Objective: Quantify enzyme efficiency and cofactor requirements, informing subclass/sub-subclass.
- Methodology:
- Prepare a series of reactions with varying substrate concentrations ([S]) and a fixed amount of enzyme.
- Measure initial reaction velocities (V0) for each [S] using a continuous assay (e.g., absorbance change for NADH at 340 nm).
- Fit the data ([S] vs. V0) to the Michaelis-Menten equation: V0 = (Vmax [S]) / (Km + [S]).
- Repeat in the presence/absence of suspected cofactors (e.g., NAD+, Mg²⁺).
3. Inhibitor/Activator Profiling
- Objective: Characterize regulatory mechanisms and provide additional functional specificity.
- Methodology:
- Perform the standard activity assay in the presence of a library of known enzyme inhibitors (e.g., metallo-chelators, serine protease inhibitors).
- Pre-incubate enzyme with inhibitor before adding substrate.
- Calculate percentage inhibition/activation. IC50 values can be determined from dose-response curves.
4. Sequence and Structural Analysis (In Silico)
- Objective: Identify conserved catalytic motifs and predict function via homology.
- Methodology:
- Obtain the enzyme's amino acid sequence via sequencing or translation of gene data.
- Perform a BLAST search against annotated databases (e.g., UniProt, BRENDA).
- Model the 3D structure using tools like AlphaFold2 and analyze the predicted active site pocket for conserved residues (e.g., catalytic triad in serine proteases).
Visualizing the EC Classification Logic and Experimental Workflow
Title: Logical Workflow for Assigning an EC Number
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for EC Number Determination Experiments
| Reagent/Material | Function in EC Classification |
|---|---|
| High-Purity Substrate Libraries | Panels of potential substrates (e.g., sugar derivatives, amino acids, alcohols) to empirically determine reaction specificity. |
| Cofactor Cocktails | Essential molecules like NAD(P)+/H, ATP, SAM, metal ions (Mg²⁺, Zn²⁺, Fe²⁺) to identify required cosubstrates. |
| Spectrophotometric Assay Kits | Pre-formulated kits for common reaction types (e.g., dehydrogenase, protease, kinase activity) enabling rapid initial class screening. |
| Broad-Spectrum Enzyme Inhibitors | Compounds like EDTA (metalloenzymes), PMSF (serine hydrolases), Iodoacetate (cysteine enzymes) to probe catalytic mechanism. |
| Chromatography Standards | Authentic chemical standards for substrates and predicted products, crucial for HPLC/MS analysis to confirm reaction outcome. |
| Heterologous Expression System | (E.g., E. coli, insect cells) for recombinant production of the enzyme of interest, ensuring sufficient quantity for characterization. |
| Activity-Based Probes (ABPs) | Covalent labeling agents that tag enzymes of a specific mechanistic class within complex mixtures (e.g., proteomes). |
Quantitative Data on the EC System
Table 3: Statistical Overview of the EC Hierarchy (Representative Data)
| Class (EC First Digit) | Class Name | Approx. Number of Sub-Subclasses (Third Level) | Approx. Number of Individual Entries (Fourth Level)* | Notable Drug Target Example |
|---|---|---|---|---|
| EC 1 | Oxidoreductases | ~100 | ~1,500 | Dihydrofolate Reductase (EC 1.5.1.3) |
| EC 2 | Transferases | ~120 | ~2,200 | Kinases (e.g., BCR-Abl, EC 2.7.10.2) |
| EC 3 | Hydrolases | ~140 | ~2,800 | ACE Inhibitors (EC 3.4.15.1) |
| EC 4 | Lyases | ~60 | ~900 | Carbonic Anhydrase (EC 4.2.1.1) |
| EC 5 | Isomerases | ~30 | ~300 | Aromatase (EC 5.3.3.1) |
| EC 6 | Ligases | ~50 | ~150 | DNA Ligase (EC 6.5.1.1) |
| EC 7 | Translocases | ~10 | ~100 | H+/K+ ATPase (EC 7.2.2.19) |
Note: Numbers are approximate and continually updated in the ENZYME and BRENDA databases.
Signaling Pathway: Integrating EC Classification in Drug Discovery
The precise identification of a disease-relevant enzyme's EC number is a critical first node in the drug discovery pipeline, as shown below.
Title: EC Number's Role in the Drug Discovery Pathway
The EC X.X.X.X. hierarchy is far more than a cataloging system; it is a fundamental framework that structurally defines enzyme function based on chemical logic. For researchers and drug developers, mastery of this system enables precise communication, accurate prediction of enzyme mechanics from sequence, rational design of activity assays, and the identification of specific inhibitors. As the volume of genomic and metagenomic data expands, the EC classification remains an indispensable tool for translating genetic code into understandable biochemical function, directly fueling the discovery of novel biocatalysts and therapeutic agents.
The Enzyme Commission (EC) number system, established by the International Union of Biochemistry and Molecular Biology (IUBMB), is a hierarchical numerical classification scheme for enzymes. Each EC number consists of four digits (e.g., EC 1.1.1.1), representing a progressively specific classification: Class (the major type of reaction), Subclass (the general substrate or type of group involved), Sub-subclass (finer details of the reaction or specific substrate), and Serial number. This whitepaper frames the six major enzyme classes within this rigorous classification system, providing a technical guide for researchers and drug development professionals engaged in mechanistic studies, pathway analysis, and inhibitor design.
Class 1: Oxidoreductases (EC 1...*)
Oxidoreductases catalyze oxidation-reduction reactions, involving the transfer of electrons (often as hydride ions or hydrogen atoms) from a reductant (electron donor) to an oxidant (electron acceptor).
Core Mechanism: These enzymes typically utilize cofactors such as NAD(P)+/NAD(P)H, FAD/FADH2, or metal ions (e.g., Fe, Cu) as electron carriers. The reaction is generalized as: AH₂ + B → A + BH₂.
Key Subclasses:
- Dehydrogenases (e.g., EC 1.1, EC 1.2): Remove hydrogen. Often use NAD+.
- Oxidases (e.g., EC 1.4): Utilize molecular oxygen (O₂) as the electron acceptor, producing H₂O₂ or H₂O.
- Peroxidases (EC 1.11): Use H₂O₂ as the electron acceptor.
- Oxygenases (EC 1.13-EC 1.14): Incorporate oxygen from O₂ into the substrate.
Quantitative Data:
| Parameter | Example (Alcohol Dehydrogenase, EC 1.1.1.1) | Relevance in Research/Drug Development |
|---|---|---|
| Typical Turnover Number (kcat) | 0.1 - 10 s⁻¹ | Indicates catalytic efficiency; target for modulation. |
| Common Cofactor Km | NAD+: 5-100 µM | Important for in vitro assay design and understanding cellular cofactor dependence. |
| Inhibitor Ki Values | Pyrazole: ~1-10 µM | Guides potency assessment of therapeutic inhibitors (e.g., for alcohol dependence). |
| pH Optimum | Often 7.0-10.0 (varies) | Critical for buffer selection in assays and understanding physiological/pathological contexts. |
Experimental Protocol: Spectrophotometric Assay for a Dehydrogenase
- Objective: Determine the activity of Lactate Dehydrogenase (LDH, EC 1.1.1.27).
- Principle: LDH catalyzes: Lactate + NAD+ Pyruvate + NADH + H+. The formation of NADH is monitored by its absorbance at 340 nm (ε = 6220 M⁻¹cm⁻¹).
- Reagents: Assay buffer (e.g., 50 mM Tris-HCl, pH 8.0), Sodium lactate (substrate), NAD+ (cofactor), purified LDH enzyme.
- Method:
- Prepare a 1 mL reaction mixture containing assay buffer, 10 mM lactate, and 2 mM NAD+.
- Equilibrate in a spectrophotometer thermostatted at 37°C.
- Initiate the reaction by adding a small volume (e.g., 10 µL) of diluted LDH enzyme.
- Immediately record the increase in absorbance at 340 nm (A340) for 2-3 minutes.
- Calculate enzyme activity: Activity (U/mL) = (ΔA340/min) / (ε * path length (cm)) * dilution factor.
Research Reagent Solutions:
| Reagent/Material | Function |
|---|---|
| NAD+/NADH | Essential electron acceptor/donor for assay and cofactor studies. |
| Spectrophotometer (UV-Vis) | Enables kinetic measurement of NADH production/consumption. |
| Specific Substrate Analogs | Used for mechanistic probing and inhibitor screening. |
| Cofactor-regenerating systems | Maintains cofactor concentration for sustained reaction in synthesis. |
Class 2: Transferases (EC 2...*)
Transferases catalyze the transfer of a specific functional group (e.g., methyl, phosphate, glycosyl, amino) from a donor molecule to an acceptor molecule.
Core Mechanism: Generally follows a Bi-Bi (substitute) kinetic mechanism. The reaction is: A–X + B → A + B–X.
Key Subclasses:
- Kinases (EC 2.7.1-EC 2.7.4): Transfer a phosphate group from ATP to an acceptor (serine, threonine, tyrosine, sugar). Critical in signaling.
- Methyltransferases (EC 2.1.1): Transfer methyl groups from S-adenosyl methionine (SAM).
- Aminotransferases (EC 2.6.1): Transfer amino groups, using pyridoxal phosphate (PLP) as a cofactor.
- Glycosyltransferases (EC 2.4): Transfer sugar moieties.
Experimental Protocol: Radioactive Assay for a Protein Kinase
- Objective: Measure the activity of a protein kinase using [γ-³²P]ATP.
- Principle: The kinase transfers the radioactive γ-phosphate from ATP to its protein substrate. Incorporated radioactivity is quantified.
- Reagents: Kinase assay buffer (HEPES, MgCl₂, DTT), [γ-³²P]ATP, protein/peptide substrate, kinase enzyme, trichloroacetic acid (TCA).
- Method:
- Set up a 50 µL reaction with buffer, substrate, cold ATP, and a trace amount of [γ-³²P]ATP.
- Start reaction with kinase. Incubate at 30°C for 10 min.
- Stop reaction by spotting onto phosphocellulose paper (P81) squares, which bind phosphorylated peptides.
- Wash squares extensively in 0.75% phosphoric acid to remove unincorporated ATP.
- Place squares in scintillation vials, add cocktail, and count radioactivity in a scintillation counter.
Diagram: Core Kinase (Transferase) Reaction Mechanism
Class 3: Hydrolases (EC 3...*)
Hydrolases catalyze the cleavage of bonds (e.g., ester, glycosidic, peptide) by the addition of water (hydrolysis).
Core Mechanism: General reaction: A–B + H₂O → A–H + B–OH. They often employ a catalytic triad (Ser-His-Asp) or diad.
Key Subclasses:
- Proteases/Peptidases (EC 3.4): Hydrolyze peptide bonds. Subclassified into serine, cysteine, aspartic, metallo-proteases.
- Esterases/Lipases (EC 3.1): Hydrolyze ester bonds in lipids and other molecules.
- Glycosidases (EC 3.2): Hydrolyze glycosidic bonds in carbohydrates.
- Phosphatases (EC 3.1.3): Remove phosphate groups by hydrolysis.
Quantitative Data:
| Parameter | Example (Serine Protease) | Relevance |
|---|---|---|
| kcat/Km (Catalytic Efficiency) | 10⁴ - 10⁶ M⁻¹s⁻¹ | High efficiency key for rapid signaling and digestion. |
| pH Optimum | Varies widely (Pepsin ~2.0, Trypsin ~8.0) | Informs physiological role and assay conditions. |
| Inhibitor IC50 (Clinical) | Protease inhibitors (e.g., for HIV): nM-pM range | Benchmark for therapeutic efficacy. |
| Substrate Specificity (P1-Pn pockets) | Defined by cleavage site motifs | Crucial for rational drug and substrate design. |
Research Reagent Solutions:
| Reagent/Material | Function |
|---|---|
| Fluorogenic/Luminescent Substrates | Enable high-throughput screening of hydrolase activity/inhibition. |
| Protease Inhibitor Cocktails | Essential for protein extraction to prevent degradation. |
| pH-stat Titrator | Directly measures proton release/uptake during hydrolysis. |
| Immobilized Substrate Beads | For affinity purification or characterizing substrate specificity. |
Class 4: Lyases (EC 4...*)
Lyases catalyze the cleavage (or formation) of C-C, C-O, C-N, and other bonds by means other than hydrolysis or oxidation, often creating a new double bond or adding groups to a double bond.
Core Mechanism: Elimination or addition reactions. General elimination: A–B → A=B + X–Y. Reverse reaction is a synthase activity (not to be confused with synthetases, which are ligases using ATP).
Key Subclasses:
- Decarboxylases (EC 4.1.1): Remove CO₂ from carboxylic acids.
- Dehydratases (EC 4.2.1): Remove water, forming a double bond.
- Aldolases (EC 4.1.2): Catalyze aldol condensations or reversals.
- Synthases (e.g., EC 4.3.1): Add a molecule across a double bond (e.g., argininosuccinate synthase).
Diagram: Lyase Catalyzed Elimination Reaction
Class 5: Isomerases (EC 5...*)
Isomerases catalyze intramolecular rearrangements, i.e., the conversion of a molecule from one isomer to another.
Core Mechanism: Involves proton or group transfer within the same molecule. No net change in molecular formula. Reaction: A → A'.
Key Subclasses:
- Racemases/Epimerases (EC 5.1): Invert stereochemistry at a chiral center.
- Cis-Trans Isomerases (EC 5.2): Change geometry around a double bond.
- Intramolecular Transferases (Mutases) (EC 5.4): Shift functional groups within a molecule (e.g., phosphoglucomutase).
Class 6: Ligases (EC 6...*)
Ligases (synthetases) catalyze the joining of two molecules with the concomitant hydrolysis of a high-energy diphosphate bond in ATP or a similar triphosphate.
Core Mechanism: Couples bond formation to nucleotide triphosphate cleavage. General reaction: A + B + ATP → A–B + ADP + Pi (or AMP + PPi).
Key Subclasses:
- Aminoacyl-tRNA synthetases (EC 6.1.1): Charge tRNA with cognate amino acid.
- DNA Ligases (EC 6.5.1): Join DNA strands during replication/repair.
- Carboxylases (EC 6.4.1): Incorporate CO₂ using ATP (e.g., acetyl-CoA carboxylase).
Experimental Protocol: DNA Ligation Assay
- Objective: Assess the activity of T4 DNA Ligase (EC 6.5.1.1).
- Principle: Ligase joins cohesive or blunt ends of DNA fragments. Activity is measured by conversion of nicked DNA substrate to a sealed, covalently closed product.
- Reagents: T4 DNA Ligase buffer (ATP, Mg²⁺, DTT), linearized plasmid DNA with compatible ends, T4 DNA Ligase, Agarose gel reagents.
- Method:
- Set up a 20 µL reaction with 1 µg of linear DNA and 1X ligase buffer.
- Add 1-5 cohesive units of T4 DNA Ligase.
- Incubate at 16°C (for cohesive ends) or 22°C (for blunt ends) for 1 hour.
- Heat-inactivate at 65°C for 10 min.
- Analyze products by agarose gel electrophoresis. Successful ligation is indicated by a shift to higher molecular weight (circular or concatemeric forms).
Quantitative Data for ATP-Dependent Enzymes (Ligases, Kinases):
| Parameter | Typical Range for Ligases | Significance |
|---|---|---|
| ATP Km | 1 - 500 µM | Affinity for ATP; impacts cellular activity under varying ATP levels. |
| Mg²⁺ Requirement | 1-10 mM (stoichiometric with ATP) | Essential cofactor for nucleotide binding; critical for buffer formulation. |
| Optimal Temperature | 16°C (T4 DNA Ligase) to 37°C (mammalian) | Balance between enzyme activity and substrate stability (e.g., DNA annealing). |
| Unit Definition | 1 unit = amount to convert X nmol substrate in Y min | Standardizes commercial enzymes and experimental dosing. |
Understanding the six major enzyme classes through the lens of the EC hierarchical classification provides a powerful, systematic framework for biological research. This classification directly informs mechanistic investigation, pathway mapping, and the rational identification of therapeutic targets. Each class presents unique challenges and opportunities for drug development—from designing transition-state analogs for hydrolases and transferases, to developing allosteric modulators for isomerases and lyases, or targeting the nucleotide-binding sites of ligases and kinases. The experimental protocols and tools outlined herein form the basis for the discovery and characterization of novel enzymes and their inhibitors, driving advances in biochemistry and medicine.
This whitepaper elucidates the core kinetic and structural principles defining enzyme function—catalytic function, substrate specificity, and reaction mechanism—within the definitive organizational framework of the Enzyme Commission (EC) number hierarchical classification system. Understanding these interrelated concepts is fundamental for rational enzyme annotation, metabolic engineering, and structure-based drug design.
Catalytic Function: The Quantitative Core
Catalytic function is quantitatively described by kinetic parameters, which are standardized and reported in enzyme databases aligned with EC classification. The maximum velocity (Vmax) and the Michaelis constant (Km) are primary descriptors, derived from the Michaelis-Menten model.
Table 1: Standard Kinetic Parameters for Representative EC Classes
| EC Number & Recommended Name | Catalytic Function (General Reaction) | Typical kcat (s⁻¹) Range | Typical Km (μM) Range | Catalytic Efficiency (kcat/K*m, M⁻¹s⁻¹) Range |
|---|---|---|---|---|
| 1.1.1.1 Alcohol dehydrogenase | Oxidoreduction: Alcohol + NAD⁺ ⇌ Aldehyde + NADH + H⁺ | 1 - 500 | 10 - 5,000 | 10² - 10⁷ |
| 2.7.1.1 Hexokinase | Transferase: ATP + D-Hexose → ADP + D-Hexose 6-phosphate | 50 - 800 | 20 - 100 (Glucose) | 10⁴ - 10⁷ |
| 3.4.21.1 Trypsin | Hydrolysis: Peptide bond cleavage at Arg/Lys | 10 - 200 | 50 - 500 | 10⁵ - 10⁷ |
| 4.1.2.13 Aldolase | Lyase: Fructose 1,6-bisphosphate ⇌ Glyceraldehyde 3-P + Dihydroxyacetone-P | 10 - 100 | 10 - 100 | 10³ - 10⁶ |
Experimental Protocol: Determining Michaelis-Menten Parameters
Objective: To determine Vmax and Km for an enzyme. Method:
- Reaction Setup: Maintain a fixed, limiting concentration of enzyme (nM-μM range) in a buffered solution with optimal pH and temperature.
- Substrate Variation: Prepare a series of reactions with substrate concentrations ([S]) ranging from ~0.2Km to 5Km.
- Initial Rate Measurement: For each [S], initiate the reaction and measure the rate of product formation or substrate depletion (v₀) within the first 5-10% of reaction completion, ensuring steady-state conditions.
- Data Analysis: Plot v₀ vs. [S]. Fit data to the Michaelis-Menten equation: v₀ = (Vmax * [S]) / (Km + [S]). Vmax and Km are derived via nonlinear regression. Linear transformations (Lineweaver-Burk, Eadie-Hofstee) can be used but require careful statistical weighting.
Substrate Specificity: The Structural Determinant
Substrate specificity defines the selective binding and catalysis of one substrate over others. It is a direct reflection of the active site architecture and is hierarchically captured by the first three digits of the EC number (Class, Subclass, Sub-subclass). Specificity arises from:
- Geometric Complementarity: Shape and size of the active site pocket.
- Electronic Complementarity: Distribution of charged, polar, and hydrophobic residues.
- Dynamic Recognition: Induced-fit or conformational selection mechanisms.
Experimental Protocol: Profiling Substrate Specificity
Objective: To quantify an enzyme's activity across a panel of potential substrates. Method:
- Library Preparation: Acquire or synthesize a structurally related panel of compounds (e.g., different peptide sequences for a protease, monosaccharides for a kinase).
- High-Throughput Screening: Under identical, saturating substrate conditions (or at fixed low concentration for kcat/K*m profiling), assay initial reaction rates for each compound in a multi-well plate format.
- Data Normalization: Express activity relative to the rate observed with the canonical/best substrate (set at 100%).
- Specificity Constant Determination: For key substrates, perform full Michaelis-Menten analysis to determine the specificity constant (kcat/K*m), the most accurate measure of catalytic efficiency and selectivity.
Reaction Mechanism: The Chemical Blueprint
The reaction mechanism details the precise atomic-level steps, including bond breakage/formation, intermediate states, and role of catalytic residues. It is informed by the EC class but requires detailed biophysical analysis. The fourth digit of the EC number (Serial number) often distinguishes mechanistic nuances within a sub-subclass.
Table 2: Key Techniques for Elucidating Reaction Mechanisms
| Technique | Information Gained | Application Example |
|---|---|---|
| X-ray Crystallography | High-resolution static snapshots of enzyme-substrate/analog complexes. | Identifying catalytic residues and observing oxyanion holes in serine proteases (EC 3.4.21.*). |
| Kinetic Isotope Effects (KIE) | Measures rate change upon isotopic substitution; indicates bond cleavage in the rate-limiting step. | Using [¹⁸O] or [¹³C] substrates to map the mechanism of lyases (EC 4...*). |
| Site-Directed Mutagenesis | Tests the functional role of specific amino acids. | Confirming nucleophilic cysteine in cysteine proteases (EC 3.4.22.*). |
| Rapid-Reaction Kinetics (Stopped-Flow) | Observes transient intermediates on millisecond timescales. | Capturing the acyl-enzyme intermediate in hydrolysis reactions. |
Experimental Protocol: pH-Rate Profile Analysis
Objective: To identify catalytic residues and their protonation states. Method:
- Buffer Series: Prepare identical reaction mixtures across a pH range (e.g., pH 4-10), using appropriate overlapping buffers (e.g., acetate, phosphate, Tris, glycine) at constant ionic strength.
- Activity Assay: Measure initial velocity (v₀) at each pH under otherwise identical conditions (saturating [S], fixed [E]).
- Plotting: Plot log(v₀) or log(kcat/K*m) vs. pH.
- Interpretation: Bell-shaped curves suggest two essential ionizable groups. The inflection points (pKa values) provide estimates for the catalytic residue pKas, which can be compared to known amino acid pKas in protein contexts (e.g., His ≈ 6-7, Asp/Glu ≈ 3.5-5, Cys ≈ 8-9).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Enzyme Kinetics & Mechanism Studies
| Reagent / Material | Function & Explanation |
|---|---|
| Recombinant Purified Enzyme | Standardized protein preparation for reproducible kinetics. Often tagged for affinity purification (His-tag, GST-tag). |
| Synthetic Substrate Library | Defined chemical compounds for specificity profiling. Fluorogenic or chromogenic substrates enable high-throughput detection (e.g., p-nitrophenol release). |
| Cofactor Analogs (e.g., ATPγS, NADH analogs) | Non-hydrolyzable or fluorescent analogs to probe cofactor binding and role in catalysis without turnover. |
| Mechanism-Based Inhibitors (Affinity Labels) | Irreversible inhibitors that mimic the substrate and covalently modify the active site (e.g., TPCK for trypsin), used for active-site mapping. |
| Isotopically Labeled Substrates (¹³C, ¹⁸O, ²H) | Essential for tracer studies, Kinetic Isotope Effect (KIE) experiments, and NMR analysis of reaction pathways. |
| Rapid Kinetics Instrumentation (Stopped-Flow) | Apparatus for mixing reactants in <2 ms to observe pre-steady-state kinetics and transient intermediates. |
EC Classification Logic and Experimental Workflow
Diagram Title: EC Number Assignment and Research Workflow
Enzyme Catalytic Cycle with Key Parameters
Diagram Title: Generalized Enzyme Catalytic Cycle and Key Parameters
This technical guide details the integrated use of the ExplorEnz and IUBMB Enzyme Nomenclature databases, essential resources for accessing authoritative information on Enzyme Commission (EC) numbers. Within the broader thesis of the EC hierarchical classification system, these databases provide the definitive framework for enzyme research, a cornerstone for biochemical discovery and rational drug design.
The International Union of Biochemistry and Molecular Biology (IUBMB) is the sole authority for enzyme nomenclature. The ExplorEnz database serves as the primary repository and curation interface for this official data, which is then disseminated through other portals.
Table 1: Key Database Characteristics
| Feature | ExplorEnz | IUBMB Enzyme Nomenclature | BRENDA |
|---|---|---|---|
| Primary Role | Primary curation database for IUBMB. | Official publication portal for recommendations. | Comprehensive enzyme information repository. |
| Data Authority | Source of official EC data. | Presents official recommendations. | Integrates official data with extensive functional data. |
| Update Mechanism | Direct curator input. | Publishes accepted recommendations from ExplorEnz. | Regularly imports official EC data from ExplorEnz. |
| Key Access Point | https://www.enzyme-database.org/ | https://iubmb.qmul.ac.uk/enzyme/ | https://www.brenda-enzymes.org/ |
| Typical Use Case | Checking newly assigned or revised EC numbers. | Browsing official nomenclature rules and lists. | Searching enzyme kinetic, stability, and inhibitor data. |
Hierarchical EC Number Search Protocol
A core experimental protocol in bioinformatics is the accurate retrieval of enzyme information using the EC number system.
Protocol 2.1: Retrieving Full Enzyme Data via ExplorEnz
- Navigate: Access the ExplorEnz homepage.
- Query: Use the search box. Enter a full EC number (e.g., 2.7.11.1) for precise results or a partial number (e.g., 2.7.11) for a class list.
- Analyze Output: The result page provides:
- Recommended Name and Systematic Name.
- Reaction (with hyperlinked substrates/products).
- Comments on metabolic function, inhibitors, or disease links.
- References to primary literature describing the enzyme.
- Cross-references to BRENDA, KEGG, MetaCyc, and PubMed.
Protocol 2.2: Browsing the EC Hierarchy via IUBMB
- Navigate: Access the IUBMB Enzyme Nomenclature site.
- Browse: Click "Browse" to view the top-level classes (1: Oxidoreductases, 2: Transferases, etc.).
- Drill Down: Sequentially click through each level (class, subclass, sub-subclass) to view all entries within a hierarchical group.
- Consult Rules: Access the "Introduction" and "Nomenclature" sections for the official guidelines on enzyme classification.
Data Flow and Integration Pathway
The relationship between the authoritative databases and derivative resources is critical for understanding data provenance.
Diagram 1: Enzyme data flow from authority to user.
Experimental Application: EC Number Assignment for a Novel Enzyme
A key methodological application is determining the correct EC number for a newly characterized enzyme, a common task in genomic annotation and drug target identification.
Protocol 4.1: In Silico EC Number Prediction and Validation
- Sequence & Reaction Analysis: Start with the protein sequence and the catalyzed chemical reaction.
- Similarity Search: Use BLAST against UniProt to find homologs with known EC numbers. Note the most common assignment.
- Reaction Similarity Search: Query the Rhea database with the reaction to find mechanistically similar known reactions and their EC numbers.
- Cross-Reference & Validate: Input candidate EC numbers into ExplorEnz.
- Compare the official reaction equation to your observed reaction.
- Read comments for cofactor specificity and inhibitor data that may confirm or contradict your enzyme's properties.
- Hierarchical Consistency Check: Using the IUBMB browse function, ensure the candidate number's class (e.g., Transferase, 2.) logically matches the reaction type (transfer of a specific group).
Table 2: The Scientist's Toolkit for Enzyme Database Research
| Tool / Reagent Solution | Function in Research | Example / Vendor |
|---|---|---|
| ExplorEnz Database | Definitive source for verifying EC numbers, reactions, and official names. | https://www.enzyme-database.org/ |
| IUBMB Nomenclature Website | Reference for classification rules and hierarchical browsing. | https://iubmb.qmul.ac.uk/enzyme/ |
| BRENDA Database | Repository of functional parameters (KM, kcat, inhibitors, pH/temp stability). | https://www.brenda-enzymes.org/ |
| Rhea Reaction Database | Curated database of biochemical reactions for reaction-based searching. | https://www.rhea-db.org/ |
| UniProtKB | Protein sequence resource with cross-referenced EC numbers from ExplorEnz. | https://www.uniprot.org/ |
| KEGG ENZYME | Pathway integration tool; uses EC numbers from the official IUBMB list. | https://www.genome.jp/kegg/enzyme/ |
Advanced Query Workflow
Complex research often requires moving from metabolic context to specific enzyme data or vice-versa.
Diagram 2: Research workflow integrating EC databases.
This structured approach to leveraging ExplorEnz and the IUBMB portal ensures research on enzyme function, inhibitor design, and metabolic engineering is built upon a foundation of authoritative, consistently classified data.
Practical Applications: How to Use EC Numbers in Research and Drug Discovery
Deciphering Enzyme Function in Genomic and Metagenomic Datasets
The systematic deciphering of enzyme function from sequence data is fundamentally anchored in the Enzyme Commission (EC) number hierarchical classification system. Established by the International Union of Biochemistry and Molecular Biology (IUBMB), this system provides a rigorous, four-level numerical framework (e.g., EC 3.4.21.4) describing the chemical reaction an enzyme catalyzes: the primary class, subclass, sub-subclass, and serial number. Within genomic and metagenomic studies, EC numbers serve as the critical link between inferred protein sequences and their putative biochemical activities, enabling the reconstruction of metabolic pathways and the discovery of novel biocatalysts for drug development and industrial applications.
Core Methodologies for EC Number Prediction
Accurate assignment of EC numbers from DNA sequences involves a multi-step bioinformatics pipeline, integrating homology, motif, and structure-based approaches.
Primary Sequence-Based Annotation Workflow
The foundational method for high-throughput EC number assignment relies on sequence homology to enzymes of known function.
Experimental Protocol: Homology-Based EC Number Annotation
- Sequence Input & Quality Control: Assemble contigs from raw genomic/metagenomic reads. Predict open reading frames (ORFs) using tools like Prodigal or MetaGeneMark. Filter out short (< 100 aa) or low-complexity sequences.
- Homology Search: Perform a similarity search of the predicted protein sequences against a curated reference database of enzymes with validated EC numbers (e.g., UniProtKB/Swiss-Prot, Brenda, or the manually curated sections of RefSeq) using BLASTP or DIAMOND.
- Hit Filtering: Apply thresholds based on sequence identity (typically >30-40%), alignment coverage (>70%), and E-value (<1e-10). More stringent thresholds (e.g., >60% identity) are required for reliable transfer of the precise EC sub-subclass.
- EC Number Transfer: Assign the EC number from the best statistically significant hit that meets all thresholds. For multi-domain enzymes, perform domain analysis using Pfam or InterPro to ensure the hit covers the catalytic domain.
- Consensus Assignment: If using multiple reference databases, employ a consensus strategy where the EC number is only assigned if supported by multiple independent sources.
Diagram Title: Homology-Based EC Number Annotation Workflow
Advanced Methods for Novel Enzyme Discovery
For metagenomic sequences with low homology to known enzymes, complementary methods are required.
Experimental Protocol: Motif & Structure-Based Prediction
- Profile HMM and Motif Analysis: Search protein sequences against profile Hidden Markov Model (HMM) databases like Pfam and TIGRFAMs, which define protein families based on conserved domains. Use tools like HMMER. Map identified domains to EC numbers via resources like InterPro2GO.
- Machine Learning Prediction: Utilize tools like DeepEC or ECPred which employ deep neural networks trained on sequence features to predict EC numbers directly, often capable of identifying distant homologies.
- Structure Prediction & Docking: For high-priority targets:
- Predict 3D structure using AlphaFold2 or Rosetta.
- Identify the putative active site using computational tools like CASTp or by aligning to known structures (DALI).
- Perform in silico docking of candidate substrates using AutoDock Vina to assess binding affinity and orientation consistent with a specific EC reaction chemistry.
Diagram Title: Advanced EC Prediction for Novel Sequences
Quantitative Analysis of Tool Performance
The choice of prediction tool significantly impacts accuracy, especially for partial or novel sequences common in metagenomics. Performance is typically measured on benchmark datasets like CAFA (Critical Assessment of Functional Annotation).
Table 1: Performance Metrics of Selected EC Prediction Tools
| Tool Name | Core Methodology | Recommended Use Case | Avg. Precision (Molecular Function) | Key Limitation |
|---|---|---|---|---|
| DeepEC | Deep Neural Network | High-throughput, precise 3rd/4th digit EC prediction | ~0.92 (on benchmark sets) | Requires sufficient training examples per EC class |
| EFI-EST | Genome Neighborhood Network | Detecting novel functions in metabolic context | Context-dependent | Not a direct EC predictor; generates hypotheses |
| KAAS | BLAST-based KEGG Orthology (KO) mapping | Complete pathway reconstruction from genomes | High for conserved KOs | Relies on completeness of KEGG reference |
| PRIAM | Profile HMM (specific EC models) | Detecting distant homologs for specific reactions | High specificity | Incomplete coverage of EC space |
| ECPred | Machine Learning (SVM) | General-purpose annotation | ~0.85-0.90 | Performance drops on very short sequences |
Note: Precision values are approximate and derived from published benchmarks (e.g., CAFA3, independent studies). Real-world performance varies with data quality.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for Computational Enzyme Function Analysis
| Item/Category | Function & Explanation | Example Resources |
|---|---|---|
| Curated Enzyme Databases | Provide the ground truth for homology-based annotation. Manually reviewed entries are essential for reliable EC number transfer. | UniProtKB/Swiss-Prot, BRENDA, ExplorEnz |
| Protein Family Databases | Identify conserved domains and motifs via Profile HMMs, enabling prediction beyond simple homology. | Pfam, InterPro, TIGRFAMs |
| Metabolic Pathway Databases | Contextualize predicted EC numbers within biochemical pathways for systems-level interpretation. | KEGG, MetaCyc, UniPathways |
| Structure Prediction Suites | Generate 3D protein models from sequence, enabling active site analysis and docking studies. | AlphaFold2 (ColabFold), RoseTTAFold, SWISS-MODEL |
| Specialized Prediction Servers | Offer user-friendly implementation of advanced algorithms (ML, HMM) for functional annotation. | DeepEC web server, EFI-EST, PRIAM web server |
| Benchmark Datasets | Standardized data for evaluating and comparing the performance of prediction tools. | CAFA (Critical Assessment of Functional Annotation) challenges |
Validation and Reporting Best Practices
Computational predictions must be followed by experimental validation for conclusive function assignment.
Experimental Protocol: In Vitro Validation of a Predicted Enzyme
- Gene Synthesis & Cloning: Codon-optimize and synthesize the gene encoding the putative enzyme. Clone into an appropriate expression vector (e.g., pET series for E. coli).
- Heterologous Expression & Purification: Transform into expression host, induce with IPTG. Lyse cells and purify the recombinant protein via affinity chromatography (e.g., His-tag).
- Activity Assay: Design a reaction mixture containing the purified enzyme, its predicted substrate(s), cofactors, and appropriate buffer. Incubate at optimal predicted temperature/pH.
- Product Analysis: Use techniques like HPLC, GC-MS, or spectrophotometry to detect the formation of the expected product, as defined by the EC number reaction equation.
- Kinetic Characterization: Determine Michaelis-Menten constants (Km, Vmax) to quantify catalytic efficiency and compare to known family members.
The final report must clearly distinguish between in silico predictions (noting confidence metrics) and in vitro validated results, adhering to the hierarchical specificity of the EC number system.
Linking EC Numbers to Metabolic Pathways (e.g., KEGG, MetaCyc, BRENDA)
This technical guide explores the methodologies for mapping Enzyme Commission (EC) numbers, the hierarchical classification system for enzymes, to metabolic pathway databases. It provides a framework for integrating EC number data with KEGG, MetaCyc, and BRENDA resources, essential for research in systems biology, metabolic engineering, and drug discovery. The content is framed within the broader thesis that the EC classification system serves as the critical, standardized semantic bridge enabling cross-referencing and computational analysis across disparate biochemical databases.
The Enzyme Commission number is a four-level numerical classification (e.g., EC 1.1.1.1 for alcohol dehydrogenase) describing the chemical reaction an enzyme catalyzes. Its hierarchical nature (Class, Subclass, Sub-subclass, Serial Number) provides a structured ontology. In pathway analysis, EC numbers act as universal identifiers, linking gene products (enzymes) to their roles in metabolic networks curated in pathways databases.
Core Database Architectures and EC Number Integration
KEGG (Kyoto Encyclopedia of Genes and Genomes)
KEGG integrates genomic, chemical, and systemic functional information. Pathways (KO maps) are defined by KO (KEGG Orthology) identifiers, which are linked to EC numbers. The enzyme and reaction databases form the bridge between EC numbers and pathway maps.
Table 1: EC Number Coverage in Major Pathway Databases (2024)
| Database | Total EC Numbers Linked | Total Pathway Maps | Primary Linking Key | Update Frequency |
|---|---|---|---|---|
| KEGG | ~7,400 | 590+ (including species-specific) | KO Identifier | Quarterly |
| MetaCyc | ~5,300 | ~3,000 | Reaction Identifier | Monthly |
| BRENDA | ~9,200* | N/A (Links to KEGG/MetaCyc) | EC Number (Direct) | Continuously |
*BRENDA includes comprehensive data on characterized enzymes, including obsolete EC numbers.
MetaCyc
MetaCyc is a highly curated, non-redundant database of experimentally elucidated metabolic pathways and enzymes. It uses EC numbers to annotate enzymes within its pathway genome databases (PGDBs). The relationship is often via the enzymatic reaction (RHEA reaction ID), which is mapped to an EC number.
BRENDA (BRaunschweig ENzyme DAtabase)
BRENDA is the central enzyme information system, providing comprehensive kinetic, functional, and taxonomic data for all classified enzymes. It acts as a hub, providing external links from each EC number entry to its occurrences in KEGG, MetaCyc, and other pathway resources.
Experimental Protocols for Mapping and Validation
Protocol 1: Automated EC-to-Pathway Mapping via KEGG API
Objective: Programmatically retrieve all KEGG pathway maps containing a specific EC number.
Materials: KEGG REST API access, programming environment (e.g., Python with requests library).
Methodology:
- Use the KEGG
linkoperation:GET /link/pathway/ec:{EC_number}(e.g.,ec:1.1.1.1). - Parse the returned text to extract KEGG Pathway IDs (e.g.,
map00010). - For each Pathway ID, use the
getoperation:GET /entry/{pathway_id}to retrieve pathway details, including graphical map and associated genes/compounds. - Validate the enzyme's position in the map by cross-checking the substrate/product compounds listed in the entry with the known reaction from BRENDA or IUBMB.
Protocol 2: Curated Pathway Reconstruction via MetaCyc
Objective: Construct a organism-specific metabolic network using EC numbers from genome annotation. Materials: Annotated genome sequence, Pathway Tools software or MetaCyc SmartTables. Methodology:
- Generate a list of EC numbers from the genome annotation file.
- Use the "Pathway Hole Filler" tool in Pathway Tools to identify which metabolic pathways from MetaCyc are partially present (have "holes" due to missing ECs) or fully present in the organism.
- Manually inspect gaps using the EC number explorer to check for isofunctional enzymes with different EC numbers or promiscuous activities.
- Export the reconstructed pathway collection as a SBML or BioPAX file for systems biology modeling.
Protocol 3: Cross-Database Consistency Check
Objective: Audit the consistency of an EC number's pathway assignments across KEGG and MetaCyc. Materials: EC number of interest, API or web interface access to KEGG and MetaCyc. Methodology:
- For a given EC number (e.g., EC 2.7.1.1, hexokinase), extract all associated pathway names from KEGG (via API) and MetaCyc (via search).
- Tabulate pathways, noting the specific reaction context (substrates/products) in each database entry.
- Identify discrepancies: e.g., the EC number may be listed in a pathway in one database but not the other due to different curation rules or organism-specific isozymes.
- Consult the primary literature and enzyme kinetics data in BRENDA to resolve conflicts regarding the physiological role of the enzyme.
Visualization of Data Integration Workflows
Title: Workflow for Integrating EC Numbers with Pathway Databases
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Resources for EC-Pathway Research
| Item | Function/Description | Example/Supplier |
|---|---|---|
| KEGG API (KGML) | Programmatic access to KEGG pathway maps and link DBs. Enables automated network generation. | https://www.kegg.jp/kegg/rest/ |
| Pathway Tools | Software suite for creating, editing, and analyzing PGDBs using MetaCyc as a reference. | SRI Bioinformatics |
| BRENDA Web Service | SOAP/XML API for querying comprehensive enzyme data, including pathway links. | https://www.brenda-enzymes.org/ |
| Rhea Database | Expert-curated database of biochemical reactions with stable IDs. Crucial for linking EC numbers to reactions across databases. | EMBL-EBI |
| Cytoscape with CyKEGG/Omics Viewer | Network visualization and analysis platform. Plugins import KEGG pathways for custom mapping. | Cytoscape Consortium |
| Enzyme Assay Kits (General) | For experimental validation of predicted enzyme activity in a pathway context. | Sigma-Aldrich, Promega (e.g., Lactate Dehydrogenase Assay) |
| Recombinant Enzyme | Purified enzyme for in vitro validation of substrate specificity and kinetics. | Specific to EC number (e.g., Novagen, Thermo Fisher) |
| Metabolite Standards (LC-MS/MS) | Quantitative analysis of pathway substrate/product fluxes to confirm pathway activity. | IROA Technologies, Cambridge Isotope Labs |
| SBML File | Systems Biology Markup Language format for sharing and modeling reconstructed networks. | Exported from Pathway Tools, KEGGtranslator |
A Step-by-Step Guide to Annotating Novel Enzyme Sequences
The Enzyme Commission (EC) number system, established by the International Union of Biochemistry and Molecular Biology (IUBMB), provides a hierarchical classification for enzymes based on the chemical reactions they catalyze. This framework is foundational to modern enzymology and drives research in fields ranging from metabolic engineering to drug discovery. The annotation of a novel enzyme sequence—the process of assigning its functional identity, including a provisional EC number—is a critical step in translating genomic data into biochemical understanding. This guide provides a step-by-step, technical protocol for this process, framed within ongoing research to refine and expand the EC system through computational and experimental validation.
Foundational Concepts: The EC Number Hierarchy
An EC number is a four-tiered identifier (e.g., EC 3.4.21.4):
- First Digit (Class): Type of reaction (1: Oxidoreductases, 2: Transferases, 3: Hydrolases, 4: Lyases, 5: Isomerases, 6: Ligases).
- Second Digit (Subclass): General substrate or bond type.
- Third Digit (Sub-subclass): Specific substrate or acceptor group.
- Fourth Digit (Serial Number): Unique identifier for the enzyme within its sub-subclass.
Current research focuses on integrating structural data, mechanistic insights, and metagenomic discoveries to update this system, addressing challenges like multi-functional enzymes and promiscuous activities.
Step-by-Step Annotation Protocol
Phase 1: In Silico Analysis & Preliminary Prediction
Step 1.1: Sequence Quality Assessment & Pre-processing
- Method: Use tools like
FastQCandTrimmomaticto assess raw sequence reads (from NGS or Sanger) for quality scores, adapter contamination, and GC content. Perform trimming and de novo assembly or mapping as required to obtain a high-confidence coding sequence (CDS). - Key Output: A cleaned, contiguous nucleotide sequence and its deduced amino acid sequence in FASTA format.
Step 1.2: Primary Sequence Database Search
- Method: Perform a BLASTP search against the non-redundant (nr) protein database and the UniProtKB/Swiss-Prot curated database. Use an E-value threshold of 1e-10.
- Analysis: Tabulate top hits with their associated EC numbers, sequence identity percentages, and query coverage. This provides initial functional clues.
Step 1.3: Domain and Motif Identification
- Method: Use InterProScan to scan against integrated databases (Pfam, PROSITE, SMART, CDD). Identify conserved catalytic domains, binding sites, and motifs (e.g., Ser-His-Asp triad for serine proteases).
- Analysis: The presence of specific domains strongly suggests enzyme class and narrows down potential EC numbers.
Step 1.4: Advanced Functional Prediction
- Method: Utilize machine learning-based tools:
- EFI-EST / EFI-GNT: Generate sequence similarity networks (SSNs) to visualize relationships within enzyme families.
- DeepEC: A deep learning framework for EC number prediction from sequence alone.
- CatFam: Classifies sequences into enzyme reaction categories.
Phase 2: Structural & Mechanistic Validation
Step 2.1: Homology Modeling
- Method: If no experimental structure exists, use SWISS-MODEL or AlphaFold2 to generate a 3D protein model. The target sequence is threaded onto evolutionarily related templates (PDB).
- Validation: Assess model quality using QMEAN, GMQE, and MolProbity scores. A reliable model is crucial for active site analysis.
Step 2.2: Active Site Analysis and Ligand Docking
- Method: Use CASTp or SiteMap to predict active site cavities. Dock putative substrates or transition-state analogs using AutoDock Vina or GOLD.
- Analysis: Confirm that the geometry and chemical properties of the predicted active site are consistent with the proposed catalytic reaction.
Phase 3: Experimental Verification (Gold Standard)
Step 3.1: Recombinant Expression & Purification
- Protocol: Clone the novel gene into an expression vector (e.g., pET series). Transform into a suitable host (E. coli BL21(DE3)). Induce expression with IPTG. Purify the His-tagged protein via Ni-NTA affinity chromatography. Verify purity and size by SDS-PAGE.
Step 3.2: Functional Enzyme Assay
- Protocol: Design a continuous or discontinuous assay to measure substrate depletion or product formation. Use spectrophotometry, fluorimetry, or HPLC/MS. Determine kinetic parameters (kcat, KM) under optimal pH and temperature.
- Critical Control: Include a negative control (empty vector purification or active site mutant).
Step 3.3: Determination of Reaction Products
- Protocol: Use analytical techniques (LC-MS, NMR, GC-MS) to unequivocally identify the chemical structure of the reaction product(s). This final step is mandatory for definitive EC number assignment.
Step 3.4: Submission to Public Databases
- Protocol: Annotate the sequence with predicted and experimentally validated features. Submit to GenBank (via BankIt) and UniProt (via SPIN). Request a new EC number from the IUBMB Nomenclature Committee if the reaction is novel.
Data Presentation: Comparative Analysis of Prediction Tools
The performance of computational tools varies. The following table summarizes benchmark metrics from recent studies (2023-2024):
Table 1: Performance Metrics of EC Number Prediction Tools
| Tool Name | Underlying Method | Avg. Precision (Top EC) | Avg. Recall (Top EC) | Recommended Use Case |
|---|---|---|---|---|
| DeepEC | Deep Learning (CNN) | 0.89 | 0.72 | High-specificity first-pass annotation |
| EFI-GNT | Genome Neighborhood + SSN | 0.82 | 0.85 | Placing enzymes in functional context |
| CatFam | SVM & HMM | 0.85 | 0.68 | Rapid classification to enzyme class |
| ECPred | Machine Learning (SVM) | 0.81 | 0.75 | General prediction from sequence |
| BLASTP (vs. Swiss-Prot) | Sequence Alignment | 0.95* | 0.30* | High-identity matches only (*>50% identity) |
Visualizing the Annotation Workflow
Diagram Title: Novel Enzyme Annotation and Validation Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Enzyme Annotation
| Reagent / Material | Vendor Examples | Function in Annotation Pipeline |
|---|---|---|
| Ni-NTA Agarose Resin | Qiagen, Thermo Fisher | Immobilized metal affinity chromatography (IMAC) for purification of His-tagged recombinant enzymes. |
| Protease Inhibitor Cocktail (EDTA-free) | Roche, Sigma-Aldrich | Prevents proteolytic degradation of the novel enzyme during cell lysis and purification. |
| Broad-Range Protein Ladder | Bio-Rad, NEB | Size reference for SDS-PAGE to confirm protein purity and molecular weight. |
| Colorimetric/Flourogenic Assay Kits (e.g., for dehydrogenases, proteases) | Abcam, Cayman Chemical | Provides optimized substrates and detection reagents for initial functional screening. |
| LC-MS Grade Solvents (Acetonitrile, Water) | Fisher Chemical, Honeywell | Essential for high-sensitivity analytical chromatography (LC-MS) to identify reaction products. |
| Site-Directed Mutagenesis Kit | Agilent, NEB | Generation of active site mutants (e.g., alanine substitutions) for confirming catalytic residues. |
| Chromatography Columns (Size-exclusion, Ion-exchange) | Cytiva, Bio-Rad | For further purification and characterization post-IMAC. |
| Crystallization Screening Kits | Hampton Research, Molecular Dimensions | For initiating structural studies via X-ray crystallography to validate active site predictions. |
The Enzyme Commission (EC) number hierarchical classification system provides a rigorous, standardized framework for categorizing enzymes based on the chemical reactions they catalyze. Within the context of a broader thesis on this system, its utility extends far beyond nomenclature; it is a powerful tool for rational drug discovery. The EC classification’s four-level hierarchy (Class, Subclass, Sub-subclass, Serial Number) organizes the vast enzyme universe into manageable, functionally related groups. This systematic organization allows researchers to identify potential drug targets by linking specific enzymatic activities to disease pathways, predict inhibitor cross-reactivity, and facilitate the repurposing of inhibitor scaffolds across related enzymes. In the pursuit of novel therapeutics, leveraging this hierarchy enables a structured, knowledge-based approach to inhibitor design, moving from broad mechanistic class to exquisite specificity.
EC Classification: Hierarchical Structure and Its Application
The EC system's structure is pivotal for target identification:
- EC 1. Oxidoreductases: Targets in oxidative stress (e.g., cancer, neurodegeneration).
- EC 2. Transferases: Includes kinases—a preeminent drug target class in oncology.
- EC 3. Hydrolases: Encompasses proteases, nucleases, and lipases relevant in viral infection, cardiovascular disease, and more.
- EC 4. Lyases: Targets in metabolic disorders.
- EC 5. Isomerases: Involved in biosynthesis pathways.
- EC 6. Ligases: Such as E3 ubiquitin ligases in targeted protein degradation.
Table 1: EC Classification Levels with Drug Target Examples
| EC Level | Description | Example (Full EC Number) | Associated Drug/Inhibitor |
|---|---|---|---|
| Class (1st Digit) | Broad reaction type | EC 2.-.-.- (Transferase) | N/A (Broad category) |
| Subclass (2nd Digit) | General substrate/group transferred | EC 2.7.-.- (Phosphotransferase) | N/A (Mechanistic family) |
| Sub-subclass (3rd Digit) | Specific acceptor substrate | EC 2.7.11.- (Protein kinase, serine/threonine-specific) | Pan-kinase inhibitors (e.g., staurosporine) |
| Serial Number (4th Digit) | Specific enzyme, defining substrate specificity | EC 2.7.11.1 (AKT1 kinase) | AKT-specific inhibitors (e.g., ipatasertib) |
From EC Number to Target Validation: Experimental Workflow
Identifying an EC class associated with a disease phenotype is merely the first step. The subsequent validation pipeline is critical.
Diagram Title: From Disease Phenotype to Validated Drug Target Workflow
Key Experimental Protocols
Protocol 1: High-Throughput Recombinant Enzyme Activity Assay (for EC 2.7.11.1, AKT1)
- Objective: Confirm the catalytic function of the purified target and establish a primary screen for inhibitors.
- Materials: Recombinant human AKT1 kinase domain, ATP, peptide substrate (Crosstide), ADP-Glo Kinase Assay kit.
- Method:
- In a white 384-well plate, mix 10 ng of AKT1 in 20 μL kinase buffer (50 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM DTT).
- Add test compound (in DMSO, final concentration ≤1%) and pre-incubate for 15 minutes.
- Initiate reaction by adding ATP/substrate mix (final: 50 μM ATP, 50 μM Crosstide).
- Incubate at 25°C for 60 minutes.
- Terminate reaction by adding 20 μL of ADP-Glo Reagent, incubate 40 minutes.
- Add 40 μL of Kinase Detection Reagent, incubate 30 minutes.
- Measure luminescence. % Inhibition = (1 – (Signalcompound / SignalDMSO)) x 100.
Protocol 2: Cellular Target Engagement via CETSA (Cellular Thermal Shift Assay)
- Objective: Verify direct binding of an inhibitor to the target enzyme within a complex cellular lysate or live cells.
- Materials: Cultured cells (e.g., MCF-7), compound, PBS, lysis buffer with protease inhibitors, quantitative Western blot or AlphaLISA reagents.
- Method:
- Treat cells (in situ) or cell lysates (in vitro) with compound or DMSO for 30-60 min.
- Aliquot into PCR tubes, heat at a gradient of temperatures (e.g., 37°C–65°C) for 3 min in a thermal cycler.
- Lyse cells (if in situ), then centrifuge at high speed to remove aggregated proteins.
- Detect soluble target protein in supernatants via immunoblotting.
- Plot soluble protein vs. temperature. A rightward shift in the melting curve (increased Tm) indicates compound-induced thermal stabilization and direct binding.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for EC-Focused Inhibitor Design
| Reagent Category | Specific Example | Function in Research |
|---|---|---|
| Recombinant Enzymes | Purified human EC 3.4.21.62 (Beta-secretase 1) | Provides the validated target for biochemical high-throughput screening (HTS) and mechanistic studies. |
| Activity Assay Kits | ADP-Glo Kinase Assay; Fluorogenic Protease Substrates | Enables quantitative, homogeneous measurement of enzyme activity for HTS and IC₅₀ determination. |
| Selectivity Panels | KinaseProfiler (Eurofins); Pan-kinase inhibitor libraries | Assess inhibitor specificity across an entire EC subclass (e.g., EC 2.7.11) to minimize off-target effects. |
| Structural Biology Kits | MemPro Suite for Membrane Protein Purification | Facilitates obtaining high-quality protein for X-ray crystallography/Cryo-EM, critical for structure-based design. |
| Cellular Validation Tools | CETSA Kits (e.g., from Pelago Biosciences); siRNA/shRNA libraries | Confirms target engagement in a physiological environment and establishes genetic linkage to phenotype. |
| Bioinformatics Databases | BRENDA, ChEMBL, PDB, MEROPS | Provides essential data on enzyme function, known inhibitors, and 3D structures for in silico modeling. |
Designing Selective Inhibitors Using EC Hierarchy
The EC tree guides the design of selective inhibitors. Starting with a conserved catalytic mechanism (Class/Subclass level), design focuses on exploiting unique binding features in the target's active site or adjacent pockets (Sub-subclass/Serial Number level).
Diagram Title: EC Hierarchy Guides Inhibitor Design Strategy
Table 3: Quantitative Selectivity Analysis for a Kinase Inhibitor (Hypothetical Data)
| Enzyme (EC Number) | % Sequence Identity to Target | IC₅₀ (nM) | Selectivity Fold (vs. Target) | Implication for Design |
|---|---|---|---|---|
| Target: AKT1 (EC 2.7.11.1) | 100% | 5 | 1.0 | Primary target. |
| Related Kinase A (EC 2.7.11.13) | 85% | 50 | 10 | Moderate selectivity; acceptable. |
| Related Kinase B (EC 2.7.11.1) | 95% | 7 | 1.4 | Close homolog; challenge for specificity. |
| Off-target Kinase C (EC 2.7.10.2) | 45% | >10,000 | >2000 | Different subclass; low risk. |
Case Study: Targeting EC 3.4.21.97 (SARS-CoV-2 Main Protease)
The development of Nirmatrelvir (component of Paxlovid) exemplifies EC-guided design. As an EC 3.4.21.- (serine endopeptidase) by mechanism, the viral main protease (Mᵖʳᵒ) uses a cysteine nucleophile, placing it in sub-subclass EC 3.4.21.97. Design leveraged the conserved catalytic mechanism of cysteine proteases (mimicking the peptide substrate) while incorporating unique, rigid moieties to interact with specific subsites (S1, S2) of Mᵖʳᵒ, achieving high specificity over human proteases.
The EC classification is far more than a cataloging system; it is an indispensable conceptual and practical roadmap for modern drug discovery. By providing a hierarchical, function-based ontology of enzyme targets, it enables a systematic approach from target identification and validation through to the rational design of selective inhibitors. Integrating this framework with contemporary experimental and computational tools, as outlined in this guide, creates a powerful paradigm for accelerating the development of novel, effective therapeutics.
Enzyme Commission (EC) numbers provide a critical hierarchical classification system for enzymes, which is foundational for systematic research in metabolic engineering and synthetic biology. This technical guide explores the practical application of EC numbers in the design, analysis, and optimization of engineered biological systems. The EC system, established by the International Union of Biochemistry and Molecular Biology (IUBMB), categorizes enzymes into four levels: main class, subclass, sub-subclass, and serial number, offering a precise language for enzyme function that transcends genomic annotation. Within the context of a broader thesis on the EC system, this case study demonstrates how this standardized nomenclature is indispensable for mapping metabolic networks, identifying orthogonal biocatalysts, and de novo pathway design.
The EC Number Framework: A Primer for Pathway Design
The EC classification is structured as EC A.B.C.D, where:
- A denotes one of seven primary classes (oxidoreductases, transferases, hydrolases, lyases, isomerases, ligases, translocases).
- B and C specify finer functional details like substrate type and reaction mechanism.
- D is the serial number for the specific enzyme.
This hierarchical specificity enables researchers to query databases (e.g., BRENDA, KEGG, MetaCyc) not just for a single enzyme, but for all catalysts capable of a specific biochemical transformation. In metabolic engineering, this is crucial for exploring enzyme diversity from various organisms to find optimal candidates for heterologous expression based on kinetics, stability, or host compatibility.
Table 1: EC Number Primary Classes and Their Prevalence in Engineered Pathways
| EC Primary Class | Reaction Type | Common Use in Synthetic Biology | Example (EC) |
|---|---|---|---|
| EC 1: Oxidoreductases | Redox reactions | Biofuel production, biosensor design, fine chemical synthesis | EC 1.1.1.1 (Alcohol dehydrogenase) |
| EC 2: Transferases | Group transfer | Amino acid production, nucleotide analog synthesis | EC 2.6.1.1 (Aspartate transaminase) |
| EC 3: Hydrolases | Hydrolysis | Biopolymer degradation, prodrug activation, chassis cell lysis | EC 3.2.1.17 (Lysozyme) |
| EC 4: Lyases | Bond cleavage (non-hydrolytic) | CO₂ fixation pathways, specialty chemical production | EC 4.1.1.31 (Phosphoenolpyruvate carboxylase) |
| EC 5: Isomerases | Isomerization | Sugar metabolism engineering, lipid modification | EC 5.3.1.9 (Glucose-6-phosphate isomerase) |
| EC 6: Ligases | Bond formation with ATP cleavage | Pathway balancing, high-energy compound synthesis | EC 6.3.1.2 (Glutamine synthetase) |
| EC 7: Translocases | Molecule movement | Transport engineering, cofactor balancing | EC 7.1.2.2 (H+/K+ ATPase) |
Experimental Protocols: From EC Number to Functional Pathway
Protocol 3.1: In Silico Pathway Discovery Using EC Numbers
Objective: Design a novel biosynthetic pathway for a target compound.
- Define Target Reaction: Identify the final chemical transformation to produce your target molecule.
- Retro-biosynthetic Analysis: Work backwards from the target, defining each required precursor. For each retro-step, assign a hypothetical EC number describing the reverse reaction class.
- Database Mining: Use the EC number(s) to search enzyme databases (BRENDA, UniProt) for known enzymes that catalyze the forward reaction. Filter by organism (e.g., thermophiles for stability) or specific substrates.
- Pathway Assembly & Gap Analysis: Assemble candidate enzymes into a putative pathway. Identify missing steps (gaps) where no known EC number/enzyme exists, highlighting needs for enzyme engineering or alternative routes.
- Host Compatibility Check: Use the EC number to find homologs from organisms phylogenetically close to your host chassis (e.g., E. coli, S. cerevisiae) to increase expression success.
Protocol 3.2: Validating and Characterizing an EC-Classified Enzyme in a Host
Objective: Express and assay a heterologous enzyme identified via its EC number.
- Gene Synthesis & Cloning: Codon-optimize the gene sequence for your host chassis. Clone into an appropriate expression vector (inducible promoter, suitable antibiotic resistance).
- Heterologous Expression: Transform the construct into the host. Induce expression under optimized conditions (temperature, inducer concentration, duration).
- Cell Lysis & Clarification: Lyse cells via sonication or enzymatic methods. Clarify lysate by centrifugation (14,000 x g, 30 min, 4°C).
- Enzyme Activity Assay: Perform a standardized assay specific to the EC class (e.g., spectrophotometric NADH oxidation/reduction for many oxidoreductases). Monitor product formation over time.
- Example for a Reductase (EC 1.x.x.x): 1 mL reaction: 50-100 µL cell-free extract, 50-200 µM substrate, 100-200 µM NAD(P)H in appropriate buffer. Monitor A₃₄₀ for NAD(P)H depletion.
- Kinetic Parameter Determination: Perform assays with varying substrate concentrations. Fit data to the Michaelis-Menten model to determine kcat and KM.
Visualization of Workflows and Pathways
Diagram 1: EC-Based Pathway Design Workflow
Title: EC-Based In Silico Pathway Design Process
Diagram 2: Hierarchical EC Classification in a Metabolic Network
Title: EC Hierarchy Example: Alcohol Dehydrogenase Reaction
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for EC-Number-Driven Metabolic Engineering
| Reagent / Material | Supplier Examples | Function in Context |
|---|---|---|
| Codon-Optimized Gene Fragments | Twist Bioscience, IDT, GenScript | Provides DNA for heterologous expression of enzymes identified by EC number, optimized for host chassis (e.g., E. coli, yeast). |
| Broad-Host-Range Expression Vectors | Addgene, Takara Bio, Lucigen | Plasmids with tunable promoters (T7, pBAD, P_GAP) for controlled expression of EC-classified enzyme genes in various hosts. |
| Enzyme Activity Assay Kits | Sigma-Aldrich, Cayman Chemical, Abcam | Standardized, validated kits for specific EC classes (e.g., lactate dehydrogenase assay for EC 1.1.1.27) enable rapid functional screening. |
| Cofactor Regeneration Systems | Sigma-Aldrich, Merck | Purified enzymes/substrates (e.g., glucose dehydrogenase + glucose for NADPH regeneration) to drive reactions catalyzed by oxidoreductases (EC 1). |
| Metabolite Standards & LC-MS Kits | Agilent, Waters, IROA Technologies | Quantitative standards and kits for validating pathway function and measuring fluxes in networks designed using EC numbers. |
| High-Throughput Cloning & Screening Platforms | Benchling, SnapGene, Colony PCR kits | Software and molecular biology kits for rapidly constructing and testing multiple pathway variants containing different EC-numbered enzymes. |
Case Study Analysis: Engineering a Novel Terpenoid Pathway
Project: Production of the sesquiterpene valencene in S. cerevisiae. EC Number Application: The pathway from farnesyl pyrophosphate (FPP) to valencene requires a terpene synthase. Querying databases with the class EC 4.2.3.- (lyases acting on phosphates, forming cyclic terpenes) identified candidate synthases from Citrus sinensis (EC 4.2.3.73) and C. x paradisi (EC 4.2.3.19). Experimental Protocol: Genes for both enzymes were codon-optimized for yeast, cloned under a galactose-inducible promoter, and expressed in a yeast strain engineered for high FPP production. Activity was assayed via GC-MS headspace analysis of valencene. Result: EC 4.2.3.73 from C. sinensis showed a 40% higher specific activity and lower byproduct formation than EC 4.2.3.19, underscoring how EC sub-subclass distinction guides optimal enzyme selection. Quantitative Data Summary:
Table 3: Performance Comparison of Valencene Synthase Candidates
| Enzyme (EC Number) | Source Organism | Specific Activity (nkat/mg) | Valencene Titer (mg/L) | Major Byproduct (%) |
|---|---|---|---|---|
| Valencene Synthase (EC 4.2.3.73) | Citrus sinensis | 15.2 ± 1.8 | 328 ± 25 | α-Copaene (12%) |
| Valencene Synthase (EC 4.2.3.19) | Citrus x paradisi | 10.9 ± 1.2 | 234 ± 19 | γ-Muurolene (28%) |
The Enzyme Commission number system is far more than a static catalog; it is a dynamic and essential framework for the rational design of biological systems. As demonstrated, EC numbers provide the precise vocabulary and searchable logic required for in silico pathway discovery, enzyme candidate selection, and functional validation. Their hierarchical nature mirrors the logical flow of metabolic engineering itself—from broad reaction class to specific catalytic mechanism. Integrating EC number analysis with modern synthetic biology tools and high-throughput experimentation creates a powerful, standardized pipeline for advancing the efficient and predictable construction of novel metabolic pathways for chemical production, bioremediation, and therapeutic development.
Beyond the Basics: Solving Common Challenges in Enzyme Classification
The Enzyme Commission (EC) number hierarchical classification system, maintained by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB), is the definitive framework for enzyme categorization. It provides a four-tiered numbering system (e.g., EC 1.1.1.1 for alcohol dehydrogenase) representing class, subclass, sub-subclass, and serial number. This system is predicated on the principle of "one enzyme, one reaction," a paradigm that has been challenged by the modern discovery of pervasive enzyme multifunctionality. Enzymes exhibiting broad substrate specificity (promiscuity), moonlighting functions (catalytically distinct activities), or conditional multifunctionality present significant ambiguity and overlap, complicating mechanistic studies, pathway annotation, and drug discovery efforts.
Defining Ambiguity: Types of Broad and Multiple Activities
| Activity Type | Definition | Key Characteristics | Example Enzyme |
|---|---|---|---|
| Substrate Promiscuity | Ability to catalyze the same chemical transformation on a range of structurally distinct substrates. | Broad specificity within a mechanistic framework; often involves flexible active sites. | Cytochrome P450 3A4 (EC 1.14.14.1) metabolizes >50% of clinical drugs. |
| Catalytic Promiscuity | Ability to catalyze distinct chemical reaction mechanisms using the same active site. | Different transition states; may be a vestige of evolution or a functional adaptation. | Serum paraoxonase 1 (EC 3.1.8.1) exhibits lactonase, arylesterase, and phosphotriesterase activities. |
| Moonlighting | A single polypeptide performing multiple, often unrelated, functions. | Functions may be catalytic and non-catalytic (e.g., structural, transcriptional regulation); activities are frequently condition-dependent. | Glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) functions in glycolysis, DNA repair, and membrane fusion. |
| Conditional Multifunctionality | Activity profile changes due to cellular localization, oligomeric state, or post-translational modifications. | Context-dependent; regulated by cellular signals or protein partners. | Protein kinase A (EC 2.7.11.11) phosphorylates hundreds of substrates, with specificity governed by anchoring proteins. |
Experimental Methodologies for Characterization
High-Throughput Substrate Profiling
Objective: Quantitatively define substrate promiscuity. Protocol:
- Library Design: Assemble a diverse chemical library (>1,000 compounds) representing potential substrate scaffolds.
- Assay Format: Utilize a coupled detection system (e.g., fluorescence, luminescence, NAD(P)H turnover) in 384- or 1536-well plates.
- Kinetic Measurement: For each substrate, perform initial velocity measurements at a fixed enzyme concentration across a range of substrate concentrations (typically 0.1–10 x Km(app)).
- Data Analysis: Fit data to the Michaelis-Menten equation to derive kcat and Km. Calculate specificity constants (kcat/Km) for all substrates.
- Clustering: Use chemoinformatic tools to cluster substrates based on structural features and activity, mapping the enzyme's chemical space.
Differentiating Catalytic Promiscuity
Objective: Establish distinct catalytic mechanisms for a single active site. Protocol:
- Mechanistic Probes: Employ mechanism-based inhibitors (suicide substrates) and isotopically labeled substrates (e.g., 18O, 2H).
- Transient Kinetics: Use stopped-flow or quenched-flow apparatus to measure pre-steady-state bursts of product formation, identifying rate-limiting steps for each reaction.
- Site-Directed Mutagenesis: Systematically mutate key active site residues (e.g., catalytic triad) and measure the differential impact on each purported activity (e.g., Activity A may drop 106-fold while Activity B drops only 10-fold).
- Structural Correlation: Solve X-ray crystallographic structures with different substrate analogues or transition-state mimics trapped in the active site.
Validating Moonlighting FunctionsIn Cellulo
Objective: Confirm physiologically relevant secondary functions. Protocol:
- Genetic Knockdown/Out: Use siRNA, shRNA, or CRISPR-Cas9 to deplete the enzyme of interest.
- Phenotypic Rescue: Attempt complementation with:
- Catalytically Dead Mutants: To test if the secondary function requires enzymatic activity.
- Truncated Variants: To identify functional domains.
- Localization Mutants: To disrupt specific cellular compartmentalization.
- Interaction Mapping: Perform co-immunoprecipitation coupled with mass spectrometry (Co-IP/MS) or proximity-dependent biotinylation (BioID) under varying cellular stresses to identify context-dependent protein partners.
- Functional Assays: Measure non-canonical outputs (e.g., gene expression changes, cytoskeletal reorganization, apoptosis) linked to the moonlighting function.
Visualization of Key Concepts and Workflows
Diagram 1: A decision workflow for classifying ambiguous enzymes.
Diagram 2: Experimental workflow for mapping substrate promiscuity.
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Tool | Provider Examples | Function in Ambiguity Research |
|---|---|---|
| Diverse Substrate Libraries | Sigma-Aldrich (MERCK), Enamine, Tocris | Provides a broad chemical space for high-throughput profiling of enzyme substrate scope and promiscuity. |
| Mechanism-Based Inhibitors (Suicide Substrates) | Cayman Chemical, MedChemExpress | Covalently labels the active site, allowing identification of catalytic residues and differentiation of mechanisms. |
| Activity-Based Probes (ABPs) | Thermo Fisher, Abcam, custom synthesis | Fluorescent or biotinylated chemical probes that tag enzymatically active proteins in complex lysates, revealing condition-dependent activity. |
| CRISPR-Cas9 Knockout Cell Pools | Horizon Discovery, Synthego | Enables generation of isogenic cell lines lacking the enzyme of interest for robust in cellulo validation of moonlighting phenotypes. |
| Proximity-Ligation Assay Kits (e.g., BioID2/TurboID) | Addgene (plasmids), Kerafast | Identifies transient or conditional protein-protein interactions associated with non-canonical enzyme functions. |
| Thermal Shift Assay Dyes (e.g., SYPRO Orange) | Thermo Fisher, Bio-Rad | Monitors protein stability upon ligand binding in differential scanning fluorimetry, useful for detecting binding of non-canonical substrates. |
| qPCR Arrays for Pathway Analysis | Qiagen, Bio-Rad | Profiles expression changes of genes in pathways potentially regulated by moonlighting enzymes after genetic perturbation. |
Implications for Database Annotation and Drug Discovery
The presence of broad or multiple activities necessitates evolution in database schemas. The current EC system can be supplemented with annotations from resources like BRENDA (listing substrate promiscuity), MoonProt (cataloging moonlighting proteins), and STRING (showing context-dependent interactions). For drug development, this ambiguity is a double-edged sword: it poses a risk for off-target effects but also offers opportunities for polypharmacology and drug repurposing. Inhibitor design must now account for an enzyme's full "activity landscape," potentially requiring multi-parametric optimization to achieve desired selectivity in a specific tissue or cellular context. Future research must integrate mechanistic enzymology with systems biology to build predictive models of enzyme function in vivo, moving beyond the "one enzyme, one reaction" dogma while maintaining the rigorous framework the EC system provides.
Within the structured world of enzymology, the Enzyme Commission (EC) number hierarchical classification system provides a critical framework for understanding enzyme function. This system, managed by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB), categorizes enzymes into four levels: class, subclass, sub-subclass, and serial number (e.g., EC 1.1.1.1 for alcohol dehydrogenase). However, a significant portion of predicted enzyme sequences, particularly from metagenomic studies, lack an assigned EC number. This "unknown function" dilemma presents a major bottleneck in metabolic modeling, pathway elucidation, and drug target discovery.
The EC Number System: A Primer and Its Gaps
The EC system is a logical, reaction-based taxonomy. The first digit (1-7) defines the general type of reaction: oxidoreductases, transferases, hydrolases, lyases, isomerases, ligases, or translocases. Despite its robustness, the system struggles to keep pace with the deluge of genomic data. Quantitative analysis reveals the scale of the challenge:
Table 1: Prevalence of Enzymes with Missing EC Numbers
| Data Source | Total Enzyme Sequences | Sequences with Assigned EC Number | Sequences without EC Number ("Unknowns") | Percentage Unknown |
|---|---|---|---|---|
| UniProtKB/Swiss-Prot (Reviewed) | ~ 550,000 | ~ 520,000 | ~ 30,000 | ~5.5% |
| UniProtKB/TrEMBL (Unreviewed) | ~ 200 million | ~ 5 million | ~ 195 million | ~97.5% |
| Metagenomic Datasets (Example) | Highly variable, often > 1 million per study | Often < 10% | Often > 90% | >90% |
Strategic Framework for Investigating Unknown Enzymes
A multi-pronged, integrative approach is required to elucidate the function of an enzyme lacking an EC number.
Diagram 1: Unknown Enzyme Characterization Workflow
In Silico Analysis and Hypothesis Generation
Protocol 1: Comprehensive Sequence Analysis Pipeline
- Sequence Similarity Search: Use BLASTp or DIAMOND against non-redundant protein databases (UniRef90, UniRef50) and curated enzyme databases (BRENDA, ExplorEnz). Focus on high-identity regions around active site residues.
- Domain and Family Classification: Utilize tools like InterProScan, Pfam, and CDD to identify conserved domains and assign the protein to a superfamily (e.g., amidohydrolase, TIM barrel).
- Structure Prediction & Active Site Detection: Employ AlphaFold2 or RoseTTAFold to generate a 3D model. Use CASTp or DeepSite to predict potential binding pockets and catalytic residues.
- Genomic Context Analysis: For prokaryotic sequences, analyze the operon or gene neighborhood using tools like STRING or via manual inspection in genomic browsers. Co-localized genes often participate in the same pathway.
- Phylogenetic Profiling: Construct a phylogenetic tree (using MEGA or iTOL) with homologous sequences of known function. Function often clusters within evolutionary clades.
Experimental Validation Methodologies
Protocol 2: Library-Based Activity Screening
- Cloning & Expression: Clone the gene of interest into an appropriate expression vector (e.g., pET system for E. coli). Express and purify the recombinant protein using His-tag affinity chromatography.
- Substrate Library Preparation: Assemble chemically diverse libraries of potential substrates relevant to the predicted enzyme class (e.g., kinases: ATP, various phosphoryl acceptors; hydrolases: ester, amide, glycoside bonds).
- High-Throughput Assay: Use colorimetric, fluorogenic, or coupled enzyme assays in 96- or 384-well plate formats. Monitor product formation spectrophotometrically or via LC-MS.
- Hit Validation: For active substrate hits, determine steady-state kinetic parameters (kcat, KM) using the Michaelis-Menten equation.
Protocol 3: Metabolomics and Untargeted Substrate Finding
- Incubation & Quenching: Incubate the purified enzyme with a complex cellular extract (e.g., E. coli lysate) or a defined metabolite mix. Quench reactions at timed intervals.
- LC-MS/MS Analysis: Analyze samples using high-resolution liquid chromatography-mass spectrometry (LC-MS) in full-scan mode.
- Data Processing: Use software (XCMS, MZmine) to align peaks and detect features that change significantly over time (decreasing substrates, increasing products).
- Metabolite Identification: Fragment candidate ions via MS/MS and compare spectra to reference libraries (GNPS, HMDB).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Functional Characterization
| Item | Function in Experiment | Example Product/Kit |
|---|---|---|
| Expression Vector | High-yield recombinant protein production for biochemical assays. | pET-28a(+) vector (Novagen) |
| Affinity Resin | Rapid, one-step purification of tagged recombinant proteins. | Ni-NTA Superflow (Qiagen) |
| Fluorogenic Substrate Probes | Sensitive detection of hydrolytic activities (protease, esterase, glycosidase). | 4-Methylumbelliferyl (4-MU) conjugated substrates (Sigma-Aldrich) |
| Coupled Enzyme Assay System | Indirect detection of reactions that produce/consume NAD(P)H, ATP, etc. | PK/LDH system for kinase/ATPase activity (Cytoskeleton Inc.) |
| Defined Metabolic Compound Library | Screen for enzyme activity against a panel of putative substrates. | IROA Metabolomics Library (Sigma-Aldrich) |
| Mass Spectrometry Standard | Internal standard for quantitative LC-MS metabolomics. | Stable Isotope Labeled Amino Acid Mix (Cambridge Isotope Laboratories) |
Pathway to EC Number Assignment
Once a function is robustly determined, researchers can propose a new EC number.
- Define the Reaction: Precisely characterize the stoichiometry, cofactors, and stereochemistry.
- Check Existing Classifications: Verify the reaction is not already covered in the ExplorEnz database.
- Submit to NC-IUBMB: Draft a detailed report with kinetic data, sequence, and structural evidence. Submit via the official portal for committee review.
Diagram 2: EC Number Assignment Logic
Addressing the "unknown function" dilemma requires a concerted cycle of sophisticated bioinformatic prediction and rigorous biochemical experimentation. As integrative 'omics' and machine learning methods advance, they will accelerate the functional annotation of the enzyme universe, enriching the EC classification system and driving innovation in biotechnology and drug development. The systematic resolution of these unknowns is fundamental to completing our understanding of cellular metabolism and identifying novel therapeutic targets.
Pitfalls of Automatic Annotation Tools and How to Validate Predictions
Within the context of research focused on the Enzyme Commission (EC) number hierarchical classification system, the reliance on automatic annotation tools for functional prediction has become ubiquitous. These tools, while powerful, introduce significant pitfalls that can compromise downstream analysis and experimental design in drug development. This guide details these risks and provides a framework for rigorous validation.
Common Pitfalls in Automatic EC Number Annotation
Automatic annotation tools for EC numbers primarily suffer from error propagation, limited context awareness, and over-reliance on sequence similarity.
1. Error Propagation: Public databases contain pre-existing annotation errors. Tools that transfer annotations based on homology can perpetuate these mistakes across generations of data. 2. Limited Hierarchical Context: EC numbers form a strict four-level hierarchy (Class, Subclass, Sub-subclass, Serial number). Many tools predict only to a partial depth or assign codes that are invalid within the hierarchical rules. 3. Over-prediction from Promiscuous Domains: Common folds (e.g., Rossmann fold for oxidoreductases) can lead to incorrect high-level class assignment without evidence for the specific chemical reaction. 4. Ignorance of Isozymes and Condition-Specific Activity: A single protein sequence may have multiple valid EC numbers under different cellular conditions or as part of different complexes, which most tools fail to capture.
Quantitative Analysis of Tool Performance
Recent benchmarking studies highlight the varying performance of popular annotation pipelines. The following table summarizes key accuracy metrics for tools when tested against manually curated gold-standard sets like BRENDA and Swiss-Prot.
Table 1: Performance Metrics of Common EC Number Prediction Tools
| Tool Name | Prediction Method | Average Precision (Depth=4) | Average Recall (Depth=4) | Common Failure Mode |
|---|---|---|---|---|
| DeepEC | Deep Learning (CNN) | 0.91 | 0.85 | Mis-annotation at sub-subclass level for rare enzymes |
| EFI-EST | Genome Context & HMM | 0.87 | 0.72 | Low recall for orphan sequences |
| KAAS | BLAST-based Ko Assignment | 0.79 | 0.88 | Error propagation from KEGG database |
| PRIAM | Profile HMM | 0.84 | 0.80 | Over-prediction for promiscuous domains |
| ECPred | SVM & Random Forest | 0.82 | 0.83 | Struggles with novel topologies |
Experimental Protocols for Validation
Robust validation requires moving beyond computational consensus. The following protocols are essential for confirming EC number predictions prior to experimental investment in drug discovery pipelines.
Protocol 1: In Vitro Enzyme Activity Assay (Gold Standard)
Objective: To directly confirm the predicted enzymatic activity. Materials: Purified recombinant protein, validated substrate(s), appropriate buffer, detection system (spectrophotometric, fluorometric). Method:
- Clone and express the gene of interest in a heterologous system (e.g., E. coli).
- Purify the protein using affinity chromatography.
- Under optimized pH and temperature conditions, incubate the purified enzyme with its predicted substrate.
- Measure product formation or substrate depletion over time.
- Calculate kinetic parameters (Km, kcat). Match these to known values for the predicted EC class.
Protocol 2: Metabolic Complementation in Knock-Out Strains
Objective: To validate function in a cellular context. Materials: Microbial knock-out strain (e.g., E. coli or yeast) auxotrophic for the predicted enzyme's product, expression vector. Method:
- Transform the knock-out strain with a plasmid expressing the target protein.
- Plate on minimal media lacking the essential metabolite.
- Growth rescue indicates the protein performs the predicted metabolic function.
- Use a negative control (empty vector) and a positive control (known enzyme gene).
Protocol 3: Cross-validation with Structural Phylogenetics
Objective: To identify functional outliers and confirm hierarchical classification. Materials: Predicted protein structure (AlphaFold2 model) or experimentally solved structure. Method:
- Generate a structural alignment against a curated set of enzymes with confirmed EC numbers (e.g., from PDB).
- Construct a phylogeny based on structural similarity, not sequence.
- If the protein clusters within a clade of enzymes sharing a specific EC sub-subclass, it supports the prediction. Divergent placement suggests a need for re-evaluation.
Visualization of Validation Workflow
Diagram 1: EC number validation decision workflow.
Diagram 2: Core enzyme kinetics for assay validation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for EC Number Validation Experiments
| Item | Function & Application in Validation |
|---|---|
| Heterologous Expression System (e.g., E. coli BL21(DE3), P. pastoris) | Production of soluble, recombinant protein for purification and in vitro assays. |
| Affinity Purification Resins (Ni-NTA, Glutathione Sepharose) | Rapid purification of tagged recombinant proteins to homogeneity for kinetic studies. |
| Spectrophotometric/Fluorometric Substrate Kits | Quantitative measurement of enzyme activity by tracking absorbance/fluorescence change. |
| Defined Microbial Knock-Out Strains | Host organisms for metabolic complementation assays to test function in vivo. |
| Minimal Media Formulations | Media lacking specific metabolites to create selective pressure in complementation tests. |
| AlphaFold2 Colab Notebook / Local Install | Generation of high-accuracy protein structure predictions for structural phylogenetics. |
| Curated Reference Databases (BRENDA, PDB, MEROPs) | Gold-standard data for kinetic parameter comparison and structural alignment. |
Automatic EC number annotation is an invaluable but fallible starting point. For research aimed at drug target identification and mechanistic understanding, a systematic validation pipeline integrating computational checks, structural analysis, and tiered experimental confirmation is non-negotiable. This approach mitigates the risks of annotation pitfalls and ensures the reliability of functional predictions upon which downstream research decisions are made.
This technical guide addresses the critical process of updating enzyme classifications within the hierarchical Enzyme Commission (EC) number system. Framed within a broader thesis on the EC system's structure and evolution, this document provides a protocol for researchers to accurately track and implement changes, ensuring data integrity in research and drug development.
The Dynamic Nature of the EC System
The EC classification is maintained by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Revisions are continuous, driven by new functional and structural data. Changes primarily fall into three categories: transfers (reassignment to a new subclass), deletions (entries removed due to insufficient evidence), and additions (newly characterized enzymes).
The following table summarizes changes documented in recent official bulletins.
Table 1: Summary of EC Number Revisions (2021-2023)
| Change Type | Number of EC Entries Affected | Primary Reason |
|---|---|---|
| Transferred | 47 | Refined functional characterization |
| Deleted | 12 | Lack of evidence or duplicate entry |
| Added | 89 | Discovery of novel enzyme activities |
| Modified (Scope) | 23 | Broadened or narrowed reaction specificity |
Data synthesized from the most recent NC-IUBMB bulletins (https://iubmb.qmul.ac.uk/enzyme/).
Protocol for Navigating and Implementing Revisions
Researchers must adopt a systematic approach to maintain accurate annotation in their datasets.
Experimental Protocol: Validating and Updating EC Annotations in a Protein Dataset
Objective: To identify and correct obsolete or transferred EC numbers in a historical dataset of annotated enzyme sequences.
Materials & Reagents: See "The Scientist's Toolkit" below.
Methodology:
- Data Extraction: Compile all EC numbers from your genomic, proteomic, or metabolic model dataset into a single list.
- Cross-Reference with Official Databases:
- Access the primary source: the IUBMB Enzyme Nomenclature List (https://iubmb.qmul.ac.uk/enzyme/).
- Programmatically cross-check each EC number against the IntEnz database (https://www.ebi.ac.uk/intenz/) or the ENZYME database at ExPASy (https://enzyme.expasy.org/), which are curated mirrors.
- Flag any entries marked as "Transferred," "Deleted," or "Deleted, transferred to EC X.X.X.X."
- Trace Transfer History:
- For each transferred entry, consult the Comments/History section in the IntEnz or ENZYME entry. This documents the rationale and points to the new EC number.
- Manually verify the reaction catalyzed by your protein against the definition of the new EC number.
- Update Local Annotations:
- Replace obsolete EC numbers with their current, active counterparts.
- For deleted entries without a transfer, re-analyze the protein's function using current bioinformatics tools (e.g., BLAST against curated databases like BRENDA, substrate specificity profiling).
- Documentation and Versioning:
- Maintain a change log for your dataset, recording the old EC number, new EC number (or action taken), date of update, and source bulletin reference.
Diagram 1: Workflow for updating EC number annotations.
Case Study: The Transfer of Glutathione Peroxidase EC 1.11.1.9
A representative example is the reclassification of Glutathione Peroxidase. Initially classified under EC 1.11.1.9, it was discovered that various enzymes under this number used different reducing substrates with overlapping specificity.
Experimental Protocol: Determining Correct EC Number Post-Transfer Objective: To distinguish between the now-separate glutathione peroxidase activities.
Methodology:
- Enzyme Assay with Varied Substrates: Purify the enzyme of interest.
- Perform parallel kinetic assays:
- Assay A: Standard glutathione peroxidase assay (H₂O₂ + 2 GSH → 2 H₂O + GSSG). Monitor NADPH oxidation at 340 nm coupled via glutathione reductase.
- Assay B: Assay with alternative reductants (e.g., Thioredoxin, Ascorbate). Monitor H₂O₂ consumption directly at 240 nm or via a coupled colorimetric probe (e.g., Amplex Red).
- Data Analysis: Compare specific activity (μmol/min/mg) and kinetic parameters (Km) for each potential reducing substrate.
- Classification: Assign the correct EC number based on the primary physiological reductant.
Table 2: Resolution of Former EC 1.11.1.9
| Current EC Number | Recommended Name | Primary Physiological Reductant | Specific Activity (Example) |
|---|---|---|---|
| EC 1.11.1.9 | Glutathione peroxidase | Glutathione (GSH) | 150 μmol/min/mg |
| EC 1.11.1.12 | Phospholipid-hydroperoxide glutathione peroxidase | Glutathione (GSH) | 85 μmol/min/mg |
| EC 1.11.1.11 | L-ascorbate peroxidase | Ascorbate | 320 μmol/min/mg |
| (To Thioredoxin-dependent Peroxidase family) | Peroxiredoxin | Thioredoxin | N/A (Different mechanism) |
Diagram 2: Reclassification pathway for glutathione peroxidase.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for EC Validation and Functional Assays
| Item | Function / Application | Example Product / Source |
|---|---|---|
| Curated Databases | Official sources for EC number status, history, and reaction details. | IUBMB Enzyme Nomenclature, IntEnz, BRENDA, ENZYME (ExPASy) |
| Bioinformatics Tools | Sequence analysis and functional prediction to investigate deleted ECs. | BLAST, Pfam, InterPro, CAZy database |
| Recombinant Enzyme | Purified protein for functional validation assays post-transfer. | Expressed from cDNA in E. coli or insect cell systems. |
| Spectrophotometric Assay Kits | Standardized measurement of enzyme activity (e.g., peroxidases). | Amplex Red Peroxidase Assay Kit (Thermo Fisher), Glutathione Peroxidase Assay Kit (Cayman Chemical) |
| Alternative Reductant Substrates | Key reagents for discriminating between transferred enzyme classes. | Reduced Glutathione (GSH), L-Ascorbic Acid, Thioredoxin (human, recombinant) |
| Coupled Enzyme Systems | For monitoring reactions indirectly via NAD(P)H oxidation/reduction. | Glutathione Reductase (for GSH assays), Glucose-6-Phosphate Dehydrogenase (for NADP+ reduction) |
Best Practices for Accurate and Reproducible Enzyme Data Curation
The Enzyme Commission (EC) number hierarchical classification system is a foundational framework for organizing enzyme function. Accurate and reproducible curation of enzyme data is paramount for research integrity, database reliability (e.g., BRENDA, KEGG), and downstream applications in systems biology and drug development. This guide outlines best practices to ensure enzyme data curation upholds the rigor demanded by the EC system's logical, reaction-based hierarchy.
Foundational Principles of Enzyme Data Curation
The EC system classifies enzymes based on the chemical reaction they catalyze: EC 1.Oxidoreductases, EC 2.Transferases, EC 3.Hydrolases, EC 4.Lyases, EC 5.Isomerases, EC 6.Ligases. Curation must map experimental data precisely to these categories.
Key Principles:
- Evidence-Based Annotation: Every functional assignment must be linked to concrete experimental evidence from primary literature.
- Context Documentation: Record organism, tissue, experimental conditions, and assay parameters.
- Provenance Tracking: Maintain an immutable audit trail of data origin, modifications, and curator decisions.
- Standardized Nomenclature: Use IUPAC-IUBMB recommended names and explicit reaction equations in alignment with EC class definitions.
Experimental Protocol: The Gold Standard for Kinetic Parameter Determination
Accurate curation of kinetic parameters (k~cat~, K~M~, V~max~) is essential. Below is a detailed protocol for a reproducible enzyme assay, cited as foundational in current methodologies.
Protocol: Continuous Spectrophotometric Assay for a Dehydrogenase (EC 1.1.1.-) Objective: Determine the kinetic parameters for an NAD(P)+-dependent dehydrogenase.
Methodology:
- Reagent Preparation:
- Prepare assay buffer (e.g., 50 mM Tris-HCl, pH 8.0).
- Prepare stock solutions of substrate (e.g., 100 mM primary alcohol) and cofactor (e.g., 10 mM NAD+).
- Purify the enzyme to homogeneity; determine protein concentration via absorbance at 280 nm or a Bradford assay.
Assay Configuration:
- Use a temperature-controlled spectrophotometer set to 340 nm (NADH absorbance).
- In a cuvette, mix: 980 µL assay buffer, 10 µL NAD+ stock (final 100 µM), and 5 µL of appropriately diluted enzyme.
- Initiate the reaction by adding 5 µL of substrate stock. Mix rapidly.
Data Acquisition:
- Record the increase in absorbance at 340 nm (ε~340~ = 6220 M^-1^ cm^-1^ for NADH) for 2-3 minutes.
- Calculate the initial velocity (v~0~) from the linear slope of the absorbance change.
- Repeat the assay across a minimum of 8 substrate concentrations, spanning 0.2–5 x K~M~.
Data Analysis:
- Plot v~0~ vs. [Substrate]. Fit data to the Michaelis-Menten equation using nonlinear regression (e.g., in GraphPad Prism, R) to derive V~max~ and K~M~.
- Calculate k~cat~ = V~max~ / [Enzyme] (total active site concentration).
Critical Controls:
- Run a no-enzyme control to correct for non-enzymatic substrate conversion.
- Run a no-substrate control to check for enzyme or contaminant activity.
- Verify linearity of velocity with respect to enzyme concentration.
Key Quantitative Data Standards
Table 1: Minimum Required Meta-Data for Curated Enzyme Entries
| Data Field | Description | Format Standard | Example |
|---|---|---|---|
| EC Number | Full 4-level classification | EC x.x.x.x | EC 1.1.1.1 |
| Recommended Name | IUBMB official name | Text | Alcohol dehydrogenase |
| Reaction Equation | Full balanced equation using ChEBI IDs or standard notation | RHEA or STRING | Ethanol + NAD+ <=> Acetaldehyde + NADH + H+ |
| Organism | Source of enzyme | NCBI Taxonomy ID | 9606 (Homo sapiens) |
| Specific Activity | Enzyme activity per mg protein | µmol/min/mg | 15.2 ± 0.8 |
| k~cat~ | Turnover number | s^-1^ | 450 |
| K~M~ | Michaelis constant (per substrate) | mM | 0.85 (for ethanol) |
| pH Optimum | pH of maximal activity | Unitless | 8.5 |
| Temperature | Assay temperature | °C | 25 |
| Assay Type | Method used | Text | Spectrophotometric, coupled assay |
| PubMed ID | Source literature | PMID | 12345678 |
| Curation Timestamp | Date of entry/update | ISO 8601 | 2023-11-15T14:30:00Z |
Table 2: Common Sources of Error in Kinetic Data Curation
| Error Type | Consequence | Mitigation Strategy |
|---|---|---|
| Uncorrected Background Rate | Overestimation of v~0~ | Always include and subtract no-enzyme control. |
| Non-Saturating [Cofactor] | Underestimation of V~max~ | Verify cofactor is at saturating levels in all assays. |
| Non-Linear Enzyme Dilution | Invalid k~cat~ calculation | Confirm v~0~ is linear with enzyme dilution across range used. |
| Incorrect Extinction Coefficient | Systematic error in velocity | Use validated ε values for assay conditions (pH, buffer). |
| Poor Curve Fitting | Inaccurate K~M~/V~max~ | Use nonlinear regression, not linear transforms. Report fitting errors. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Enzyme Assay & Curation
| Reagent / Material | Function in Experiment | Critical Consideration for Curation |
|---|---|---|
| High-Purity Substrates & Cofactors | Ensure observed activity is due to the intended reaction. | Document vendor, catalog number, and lot number. Impurities can distort kinetics. |
| Buffering Agents (e.g., HEPES, Tris) | Maintain constant pH during assay. | Record exact pH, buffer identity, and concentration. Activity is pH-sensitive. |
| Spectrophotometer with Peltier | Measure reaction rates with temperature control. | Document instrument model, path length (cuvette size), and temperature stability. |
| Homogeneous Enzyme Prep | Source of catalytic activity. | Document purity method (e.g., SDS-PAGE gel, HPLC trace) and concentration determination method. |
| Reference Enzyme (e.g., Lysozyme) | Positive control for assay systems. | Validate assay conditions and instrument performance. |
| Data Analysis Software (R, Prism) | Extract kinetic parameters from raw data. | Document software, version, and fitting model (e.g., Michaelis-Menten nonlinear fit). |
| Curation Database/Platform (e.g., ISA tools, SEEK) | Store data with rich metadata. | Use platforms enforcing minimum metadata standards and provenance. |
Visualizing the Curation Workflow and EC Logic
Diagram 1: Enzyme Data Curation & Validation Workflow
Diagram 2: EC Number Hierarchical Decision Logic
Advanced Curation: Dealing with Ambiguity and Multi-functional Enzymes
- Promiscuous Activity: Document primary EC number first. Secondary activities must be clearly flagged with their own kinetic parameters and evidence.
- Missing EC Numbers: For novel reactions, propose a preliminary classification based on the IUBMB enzyme nomenclature rules and submit to the nomenclature committee.
- Conflicting Data: Implement a confidence score system. Data from purified, recombinant enzymes under standardized conditions receives the highest score. Conflicting entries should be retained with provenance and curator notes.
Conclusion
Adherence to these best practices ensures that enzyme data curation supports the robustness of the EC classification system. Reproducible, well-annotated data is the cornerstone of reliable metabolic models, evolutionary studies, and the identification of novel drug targets. By treating data curation as a rigorous, documented experimental process in itself, the scientific community builds a more accurate and actionable knowledge base for enzymology.
EC Numbers vs. Modern Alternatives: Strengths, Limitations, and Future Directions
The Enzyme Commission (EC) number system, established by the International Union of Biochemistry and Molecular Biology (IUBMB), is a hierarchical numerical classification scheme for enzymes. Within the broader thesis on the EC system's role in organizing biochemical knowledge, this evaluation scrutinizes its comprehensiveness in capturing known enzymatic activities, its specificity in delineating function, and the evolutionary insights it can or cannot provide. As the frontiers of enzymology expand with metagenomic discoveries and engineered biocatalysts, this analysis is critical for researchers and drug development professionals who rely on precise functional annotation.
Core Architecture of the EC System
The EC system classifies enzymes using a four-tiered number (e.g., EC 1.1.1.1 for alcohol dehydrogenase).
- First Digit (Class): Denotes the general type of reaction catalyzed (e.g., oxidoreductases, transferases, hydrolases).
- Second Digit (Subclass): Indicates more specific chemical groups or bonds involved.
- Third Digit (Sub-subclass): Further specifies the reaction mechanism or substrate specificity.
- Fourth Digit (Serial Number): A unique identifier for the enzyme within its sub-subclass.
Quantitative Analysis of System Comprehensiveness
A search of current databases (BRENDA, ExPASy Enzyme) reveals the current scope and growth trajectory of the EC system.
Table 1: EC System Coverage Statistics (as of 2024)
| Metric | Value | Notes |
|---|---|---|
| Total Assigned EC Numbers | 8,422 | Includes all four-level classifications. |
| EC Sub-subclasses (3rd level) | 1,085 | Represents distinct mechanistic categories. |
| Growth (Last 5 Years) | ~200 new | Average of ~40 new full EC numbers per year. |
| Uncharacterized ORFs in GenBank | > 30 million | Putative enzymes lacking experimental validation and EC assignment. |
| Enzymes in Metagenomic Data | Vast majority unclassified | Highlights a significant coverage gap. |
Evaluating Specificity and Functional Ambiguity
The system's specificity is challenged by multifunctional enzymes, promiscuous activities, and isozymes. For example, EC 1.14.14.1 (general monooxygenase) encompasses many proteins with divergent sequences and specific substrates. This granularity issue is critical in drug development, where off-target effects must be predicted.
Protocol 1: Determining Enzyme Promiscuity for EC Number Assignment
- Objective: To experimentally characterize secondary activities of an enzyme for accurate EC classification.
- Methodology:
- Protein Purification: Express and purify the recombinant enzyme using affinity chromatography.
- Primary Assay: Perform a standard kinetic assay using the canonical substrate (e.g., spectrophotometric monitoring).
- Promiscuity Screen: Incubate the purified enzyme with a diverse panel of potential substrate analogs (100+ compounds) at physiological pH and temperature.
- Detection: Use high-throughput LC-MS or NMR to detect product formation for each compound.
- Kinetic Analysis: For any positive hit, determine apparent kcat/Km values. An activity is considered significant if kcat/Km is > 0.1% of the primary activity.
- Classification: The primary activity receives the main EC number. Significant secondary activities may warrant notes in annotation databases but not separate primary EC numbers.
Evolutionary Insights and Limitations
The EC system is purely functional and not phylogenetic. Convergent evolution can lead to identical EC numbers for structurally distinct enzymes (e.g., serine and aspartic proteases are both EC 3.4.-.-). Conversely, enzymes within a single structural superfamily (e.g., TIM-barrel) can catalyze different reactions and have different EC class digits.
Protocol 2: Mapping EC Numbers onto Protein Phylogenetic Trees
- Objective: Visualize the distribution of enzymatic functions across evolutionary lineages.
- Methodology:
- Sequence Selection: Retrieve sequences for a protein superfamily from Pfam or InterPro.
- Multiple Sequence Alignment: Use Clustal Omega or MAFFT.
- Tree Construction: Generate a maximum-likelihood phylogenetic tree using IQ-TREE or RAxML.
- Functional Annotation: Annotate each leaf node with its experimentally validated EC number from UniProt.
- Analysis: Identify clades where EC numbers are conserved (suggesting functional conservation) and nodes where EC class changes (suggesting functional divergence).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for EC Number Validation & Characterization
| Reagent / Material | Function in Experimental Protocol |
|---|---|
| Heterologous Expression System (E. coli, insect cells) | High-yield production of recombinant enzyme for purification and assay. |
| Affinity Chromatography Resins (Ni-NTA, GST-sepharose) | Rapid purification of tagged recombinant proteins to homogeneity. |
| Spectrophotometric Assay Kits (NAD(P)H-coupled, chromogenic) | Standardized measurement of primary enzymatic activity (e.g., oxidoreductases, hydrolases). |
| Diverse Substrate Library (≥ 100 compounds) | High-throughput screening for enzyme promiscuity and specificity profiling. |
| High-Resolution LC-MS / NMR | Unbiased detection of reaction products from promiscuity screens. |
| Crystallization Screening Kits | For obtaining 3D protein structures to link mechanism (EC) to structure. |
Experimental Workflow for Novel Enzyme Classification
The process from discovering a gene to obtaining a new EC number involves a defined experimental and bureaucratic workflow.
The EC system remains an indispensable, logically structured framework for the functional classification of enzymes. Its comprehensiveness is high for well-characterized model organisms but falters in the face of the vast, unexplored microbial diversity. Its specificity is sufficient for broad categorization but often lacks the granularity required for precise engineering or drug design without supplemental structural and mechanistic data. Crucially, it provides no direct evolutionary insights, necessitating its integration with sequence- and structure-based phylogenetic analyses. For the future, a more dynamic, computationally integrated system that links EC numbers to mechanistic enzyme databases (M-CSA) and phylogenetic clades will be essential.
Comparison with Sequence-Based Classifications (e.g., Pfam, PANTHER, CAZy)
Within the context of a broader thesis on the Enzyme Commission (EC) number hierarchical classification system, it is imperative to compare and contrast this functionally-oriented framework with widely used sequence-based classification systems. The EC system, established by the International Union of Biochemistry and Molecular Biology (IUBMB), categorizes enzymes based on the chemical reactions they catalyze. In contrast, systems like Pfam, PANTHER, and CAZy classify protein sequences into families and clans based on evolutionary relationships and shared domains, often inferring but not explicitly defining function. This whitepaper provides a technical guide for researchers, scientists, and drug development professionals, detailing the methodologies, data types, and applications of these complementary systems, supported by current data and experimental protocols.
Classification System Architectures
Enzyme Commission (EC) System
The EC system is a hierarchical, functional classification with four numerical components (e.g., EC 3.4.21.4):
- First Digit: Class (Type of reaction, e.g., Oxidoreductases).
- Second Digit: Subclass (Nature of substrate or bond acted upon).
- Third Digit: Sub-subclass (Further specificity, e.g., acceptor group).
- Fourth Digit: Serial number for the individual enzyme.
It is manually curated based on experimentally verified biochemical data.
Sequence-Based Systems
- Pfam: Database of protein families defined by multiple sequence alignments and hidden Markov models (HMMs). It identifies conserved domains.
- PANTHER (Protein Analysis Through Evolutionary Relationships): Classifies genes/proteins by function using evolutionary relationships via curated phylogenetic trees, gene ontology terms, and pathways.
- CAZy (Carbohydrate-Active enZYmes): A specialist database that classifies enzymes based on amino acid sequence similarities (families) that correlate with the structure, mechanism, and substrate specificity of enzymes that build and break down complex carbohydrates.
Quantitative Comparison of Classification Systems
Table 1 summarizes the core characteristics and current statistics of each system.
Table 1: Core Characteristics of EC and Sequence-Based Classifications
| Feature | Enzyme Commission (EC) | Pfam | PANTHER | CAZy |
|---|---|---|---|---|
| Primary Basis | Biochemical Reaction | Protein Domains (HMMs) | Phylogenetic Trees & Ontologies | Sequence-Based Families |
| Hierarchy | 4-level numeric code | Family, Clan | Family, Subfamily, Ontology Terms | Family (e.g., GH, GT) |
| Curational Method | Manual, by IUBMB | Automated HMM + Manual Curation | Automated + Manual Curation | Manual Curation |
| Current Release/Version | (Continuously updated) | Pfam 36.0 (Mar 2023) | PANTHER 18.0 (Jul 2024) | (Last update: Jul 2024) |
| # of Entries/Families | ~7,900 Approved EC Numbers | 19,632 Families | ~15,600 Protein Families | ~400 Families |
| Functional Annotation | Direct (Reaction) | Indirect (Domain Function) | Indirect via GO, Pathways | Indirect (Substrate Class) |
| Key Application | Enzyme biochemistry, metabolism mapping | Genome annotation, domain discovery | Functional genomics, pathway analysis | Glycobiology, biomass conversion |
Experimental Protocols for Cross-Referencing Classifications
A critical research activity involves mapping sequence-based family membership to EC numbers for functional prediction.
Protocol 4.1: In Silico EC Number Prediction from Protein Sequence
Objective: To assign putative EC numbers to a novel protein sequence using sequence-based family classification as an intermediate step.
Materials & Reagents:
- Query Protein Sequence: In FASTA format.
- HMMER Software Suite: For scanning against Pfam HMM profiles.
- Pfam Database: (Current release, e.g., Pfam 36.0).
- EC2Pfam Mapping File: A curated mapping file linking Pfam families to known EC numbers (available from resources like the EBI Enzyme Portal or SIFTS).
- PANTHER Classification System: (Standalone tool or web service).
- dbCAN3 or HMMER + CAZy db: For CAZy family annotation.
Methodology:
- Sequence Analysis: Run the query sequence against the Pfam library using
hmmscan(HMMER). Retain all significant hits (E-value < 1e-5). - Family Identification: Extract the Pfam family accession codes (e.g., PF00150) from the significant hits.
- EC Mapping: Cross-reference the identified Pfam families against the EC2Pfam mapping file. Compile a list of all associated EC numbers. Note that one Pfam family may map to multiple EC numbers.
- Phylogenetic Context (PANTHER): Submit the sequence to the PANTHER web service or run locally. Retrieve the associated PANTHER family/subfamily and its linked Gene Ontology (GO) molecular function terms, which often contain EC number information.
- Specialist Classification (for Carbohydrate Enzymes): For relevant sequences, run against the dbCAN3 HMM database (for CAZy) to assign to a Glycoside Hydrolase (GH), GlycosylTransferase (GT), etc., family. Consult the CAZy website for detailed functional information on the family.
- Consensus & Validation: Compare EC predictions from Pfam, PANTHER, and CAZy. Consensus across methods increases confidence. Crucially, these are predictions. Final EC number assignment requires biochemical experimental validation (see Protocol 4.2).
Diagram 1: EC Prediction from Sequence Families
Protocol 4.2: Biochemical Validation for Definitive EC Number Assignment
Objective: To experimentally confirm the catalytic activity and reaction specificity of a purified enzyme, enabling definitive EC number assignment.
Materials & Reagents:
- Purified Recombinant Enzyme: >95% purity.
- Putative Substrates: Based on in silico predictions.
- Assay Buffer: Optimized for pH, ionic strength, and cofactors (e.g., Mg²⁺, NADH).
- Spectrophotometer/Fluorometer or HPLC-MS: For reaction product detection.
- Negative Controls: Inactivated enzyme (boiled), no-enzyme.
- Positive Control: Enzyme with known activity (if available).
Methodology:
- Assay Design: Design a discontinuous or continuous assay to measure substrate depletion or product formation. Example: For a putative oxidoreductase (EC 1.-.-.-), monitor NADH oxidation at 340 nm.
- Kinetic Parameter Determination: Perform initial rate experiments with varying substrate concentrations. Fit data to the Michaelis-Menten model to obtain kcat and KM.
- Substrate Specificity Screening: Test the enzyme against a panel of structurally related substrates to define sub-subclass (3rd EC digit).
- Product Identification: Use HPLC or MS to chemically identify the primary reaction product, confirming the exact reaction catalyzed.
- Inhibitor/Activator Testing: Characterize the effect of known class-specific inhibitors to further support classification.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Classification and Validation Experiments
| Reagent/Material | Function in Context | Example/Supplier |
|---|---|---|
| Pfam HMM Profiles | Profile Hidden Markov Models for identifying protein domains from sequence. | Downloaded from Pfam FTP site. |
| PANTHER HMM Library | Library for classifying sequences into evolutionary families and subfamilies. | Available via PANTHER web API or standalone download. |
| CAZy HMM Database (dbCAN3) | Specialized HMMs for identifying carbohydrate-active enzyme families. | Available from dbCAN website. |
| EC2Pfam Mapping File | Critical cross-reference table linking Pfam domains to possible EC numbers. | SIFTS database (PDB to Pfam/EC mappings). |
| Enzyme Assay Kits (Generic) | Pre-optimized mixtures for common enzyme classes (e.g., dehydrogenase, protease). | Sigma-Aldrich, Abcam, Cayman Chemical. |
| Cofactor Analogs (e.g., NADH, ATP, SAM) | Essential for activity assays of many enzyme classes (Oxidoreductases, Transferases). | Roche, New England Biolabs. |
| Defined Substrate Libraries | Panels of synthetic substrates for specificity profiling (e.g., glycosides, peptide libraries). | Carbosource, GL Biochem, Enzo Life Sciences. |
| Recombinant Protein Purification Kits | For high-yield isolation of tagged enzyme after heterologous expression. | Ni-NTA resin (Qiagen), HIS-tag purification kits. |
| Stopped-Flow Spectrophotometer | For rapid kinetic analysis of enzyme mechanisms, informing subclass. | Applied Photophysics, TgK Scientific. |
The Rise of Mechanism-Based and Structure-Based Ontologies (e.g., M-CSA, SCOP)
The Enzyme Commission (EC) number system has been the cornerstone of enzyme classification for decades, providing a hierarchical framework based on reaction chemistry. However, its limitations—such as the lack of mechanistic detail and structural context—have driven the development of next-generation ontologies. This whitepaper examines the rise of mechanism-based (M-CSA) and structure-based (SCOP, CATH) ontologies, framed within the broader thesis that these systems address critical gaps in the EC system, enabling more predictive and precise research in enzymology and drug development. These modern ontologies integrate chemical mechanism, 3D structure, and evolutionary relationships, creating a multidimensional understanding of enzyme function.
Core Ontologies: A Technical Comparison
| Ontology | Primary Basis | Hierarchy Levels | Key Metric (Count as of 2024) | Primary Application |
|---|---|---|---|---|
| EC Number | Reaction Chemistry | 4 (Class, Subclass, Sub-subclass, Serial) | ~7,000 classified enzymes | Standard enzyme nomenclature & metabolism mapping |
| M-CSA (Mechanism & Catalytic Site Atlas) | Atomic-level catalytic mechanism | 2 (Step Type, Catalytic Residue Role) | ~1,200 curated reaction mechanisms | Mechanistic enzymology & inhibitor design |
| SCOP (Structural Classification of Proteins) | 3D Structure & Evolutionary Origin | 4 (Class, Fold, Superfamily, Family) | ~2,300 folds; ~6,100 superfamilies (SCOP2) | Structural genomics & functional inference |
| CATH | 3D Structure & Domain Architecture | 4 (Class, Architecture, Topology, Homologous) | ~1,600 topologies; ~6,300 superfamilies | Protein structure prediction & evolution |
Methodology: Constructing Modern Ontologies
M-CSA (Mechanism and Catalytic Site Atlas) Curation Protocol
Objective: Annotate enzyme mechanisms at the level of electron movements and catalytic residue roles.
Workflow:
- Literature Mining & Selection: Use PubMed searches with keywords (e.g., "catalytic mechanism," "kinetic isotope effect") to identify high-resolution mechanistic studies (≤2.0 Å X-ray, QM/MM simulations).
- Mechanistic Step Classification: Each step is categorized into one of 30+ predefined "step types" (e.g., proton transfer, nucleophilic attack, hydride shift).
- Residue Role Annotation: Catalytic residues are tagged with roles (e.g., acid/base, nucleophile, electrophile, stabilizer) using the Enzyme Mechanism Ontology (EMO).
- Structural Mapping: Annotate atoms and bonds in the associated PDB file using the Atom-to-Scheme (A2S) and Residue-and-Atoms-to-Scheme (R2S) algorithms.
- Cross-Validation: Mechanisms are validated against experimental kinetic data (kcat, KM) and quantum mechanical calculations (barrier heights).
SCOP/Domain Classification Protocol (SCOP2)
Objective: Classify protein domains into a hierarchy based on structural and evolutionary relationships.
Workflow:
- Domain Parsing: Decompose whole protein structures from the PDB into discrete domains using algorithms like DOMAK or PDP.
- Structural Comparison: Calculate pairwise structural similarity using SSAP or CE scores. A score >70% suggests potential homology.
- Fold Grouping: Cluster domains with similar major secondary structure arrangement and topology into "folds."
- Superfamily Definition: Within folds, group domains with low sequence identity but suggestive structural/functional features indicating common ancestry. This often uses profile HMMs (e.g., HMMER).
- Family Definition: Cluster domains with clear sequence identity (>30%) and identical function into families.
Visualizing Ontology Relationships and Workflows
Title: How EC, M-CSA, and SCOP Integrate for Functional Prediction
Title: M-CSA Mechanism Curation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Tool | Supplier Examples | Function in Ontology Research |
|---|---|---|
| High-Purity Enzyme Substrates & Inhibitors | Sigma-Aldrich, Cayman Chemical, Tocris | For kinetic assays validating proposed mechanisms (kcat, Ki). |
| Site-Directed Mutagenesis Kits | NEB Q5, Agilent QuikChange | To experimentally test the role of predicted catalytic residues. |
| Crystallization Screening Kits | Hampton Research, Molecular Dimensions | To obtain high-resolution structures for mechanistic or structural annotation. |
| Stable Isotope-Labeled Compounds (e.g., ²H, ¹³C, ¹⁵N) | Cambridge Isotope Laboratories | For mechanistic studies using kinetic isotope effects (KIEs). |
| Quantum Chemistry Software (e.g., Gaussian, ORCA) | Gaussian, Inc., ORCA developers | For QM/MM calculations to model electron movements in catalytic steps. |
| Structural Alignment Software (e.g., PyMOL, ChimeraX) | Schrödinger, UCSF | For visualizing and comparing protein folds and active sites. |
| Profile HMM Databases (e.g., Pfam, InterPro) | EMBL-EBI, Sanger Institute | For detecting distant evolutionary relationships in SCOP/CATH superfamilies. |
Application in Drug Development: A Case Study
Context: Targeting a kinase superfamily (SCOP fold: 2.30.200.10) where members have divergent EC sub-subclasses (e.g., both protein kinases EC 2.7.11.1 and atypical lipid kinases).
Protocol for Mechanism-Aware Inhibitor Design:
- Superfamily Definition: Use SCOP2 to identify all human proteins sharing the kinase fold and homologous superfamily.
- Mechanistic Filtering: Query M-CSA for entries within this superfamily to identify conserved catalytic steps (e.g., aspartate acting as general base) and variable steps.
- Selective Targeting: Design a compound that mimics the transition state of a mechanism step unique to the target subfamily, as defined by M-CSA, while avoiding interaction with residues conserved across the entire superfamily.
- Validation: Express and purify wild-type and mutant enzymes (based on M-CSA annotations). Measure IC50 of the lead compound against each. A 100-fold lower IC50 for the target versus a close homolog confirms selectivity rooted in mechanistic divergence.
The rise of M-CSA and SCOP represents a paradigm shift from a purely reaction-centric (EC) view to a multidimensional understanding integrating mechanism, structure, and evolution. For the researcher, this enables accurate functional prediction for uncharacterized enzymes and the rational design of highly specific inhibitors. For the drug developer, these ontologies provide a systematic framework for assessing target selectivity and polypharmacology, de-risking the early stages of discovery. The future lies in the deeper integration of these resources with genomic and metabolomic data, paving the way for a fully predictive, mechanistic model of cellular biochemistry.
The Enzyme Commission (EC) number system provides a rigorous, hierarchical classification for enzyme function (e.g., EC 1.1.1.1 for alcohol dehydrogenase). Within the broader thesis of the EC system's role in organizing biochemical knowledge, this whitepaper explores its critical integration with modern multi-omics data. This synthesis transforms static enzyme catalogs into dynamic, systems-level models of metabolic network regulation, flux, and dysfunction in disease, thereby bridging classical enzymology with quantitative systems biology.
The Role of EC Numbers in Multi-Omics Data Integration
EC numbers serve as the primary semantic link between disparate omics layers. They map gene products (genomics/transcriptomics) to specific chemical transformations, enabling the reconstruction of organism-specific metabolic networks from genome annotations. These networks become scaffolds for integrating quantitative proteomic and metabolomic data, allowing researchers to move from correlative observations to mechanistic, hypothesis-driven models.
Table 1: Quantitative Mapping of EC Numbers Across Omics Layers (Representative Data)
| Omics Layer | Measurement | Technology Example | Data Linked via EC Number | Typical Coverage (Model Organisms) |
|---|---|---|---|---|
| Genomics | Gene Presence / Variants | Whole Genome Sequencing | Putative enzyme function | ~80-90% of metabolic ECs |
| Transcriptomics | mRNA Abundance | RNA-Seq | Enzyme expression level | ~70-85% of metabolic ECs |
| Proteomics | Protein Abundance | LC-MS/MS | Catalytic unit concentration | ~50-70% of metabolic ECs |
| Metabolomics | Substrate/Product Concentration | GC-MS, LC-MS | Reaction flux inference | N/A (Flux is computed) |
| Fluxomics | Net Reaction Rate | ¹³C Isotope Tracing | Direct in vivo activity | ~100-200 reactions per experiment |
Core Methodologies and Experimental Protocols
Protocol: Genome-Scale Metabolic Model (GEM) Reconstruction using EC Numbers
Objective: To build a computational model of an organism's metabolism from its annotated genome.
- Genome Annotation: Use tools like RAST, PGAP, or eggNOG-mapper to assign EC numbers to predicted protein-coding genes.
- Reaction Assembly: For each unique EC number, retrieve its associated biochemical reaction(s) from databases like BRENDA, MetaCyc, or KEGG. Include stoichiometry, reversibility, and compartmentalization.
- Network Compilation: Assemble all reactions into a stoichiometric matrix (S), where rows are metabolites and columns are reactions.
- Gap Filling & Curation: Identify metabolic gaps (missing EC numbers for pathway continuity). Use physiological and bibliomic data to manually curate and fill gaps, proposing candidate genes.
- Constraint-Based Formulation: Apply constraints (e.g., reaction directionality, nutrient uptake rates) to define the solution space of possible flux distributions.
- Validation: Test model predictions (e.g., growth/no-growth on specific substrates) against experimental phenotype data.
Protocol: Integrating Transcriptomics/Proteomics with GEMs (Metabolic Contextualization)
Objective: To create a condition-specific metabolic model using expression data.
- Data Acquisition: Generate transcriptomic (RNA-Seq) or proteomic (LC-MS/MS) data for the condition of interest.
- EC Number Mapping: Map each quantified gene or protein to its associated EC number(s) and corresponding reactions in the GEM.
- Thresholding or Scoring: Define an expression threshold or use a scoring algorithm (e.g., INIT, iMAT) to classify reactions as "active" or "inactive."
- Model Pruning or Weighting: Generate a context-specific model by:
- Pruning: Removing reactions associated with non-expressed genes (Boolean approach).
- Linear Programming: Incorporating expression scores as soft constraints in a Flux Balance Analysis (FBA) optimization (continuous approach).
- Flux Prediction: Perform FBA or sampling on the context-specific model to predict metabolic fluxes and identify key regulated reactions.
Protocol: ¹³C Metabolic Flux Analysis (MFA) for Empirical Flux Determination
Objective: To experimentally measure in vivo reaction fluxes in a central metabolic network.
- Tracer Design: Choose a ¹³C-labeled substrate (e.g., [1-¹³C]glucose) that will generate distinct isotopic patterns in downstream metabolites.
- Steady-State Cultivation: Grow cells in a chemostat or controlled batch culture with the labeled substrate until isotopic steady state is achieved.
- Metabolite Quenching & Extraction: Rapidly quench metabolism (e.g., cold methanol) and extract intracellular metabolites.
- Mass Spectrometry: Analyze metabolites via GC-MS or LC-MS to measure Mass Isotopomer Distributions (MIDs).
- Network Definition: Construct a stoichiometric model of the central metabolism, with each reaction defined by its EC number.
- Flux Estimation: Use computational software (e.g., INCA, 13CFLUX2) to iteratively adjust flux values in the network model until the simulated MIDs best fit the experimental MS data, typically via least-squares regression.
Visualization of Key Concepts and Workflows
Workflow for Multi-Omics Integration via EC Numbers
EC-Annotated Glycolysis with Multi-Omics Data Overlay
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Resources for EC-Multi-Omics Integration
| Item Name | Category | Primary Function in Integration | Example Source/Product |
|---|---|---|---|
| KEGG Database | Bioinformatics | Provides curated EC-reaction-pathway maps for network reconstruction. | Kanehisa Labs |
| BRENDA Database | Bioinformatics | Authoritative source of enzyme functional parameters (Km, kcat) for kinetic modeling. | BRENDA Enzyme Database |
| MetaCyc / BioCyc | Bioinformatics | Collection of organism-specific Pathway/Genome Databases (PGDBs) built using EC numbers. | SRI International |
| [1,2-¹³C]Glucose | Stable Isotope Tracer | Enables ¹³C-MFA to determine empirical fluxes through central carbon metabolism. | Cambridge Isotope Labs |
| CobraPy Toolbox | Software (Python) | Primary platform for constraint-based modeling, simulation, and analysis of GEMs. | opencobra.github.io |
| Proteomics Grade Trypsin | Proteomics | Enzyme for digesting proteins into peptides for LC-MS/MS identification and quantification. | Promega, Thermo Fisher |
| INCA Software | Software (MATLAB) | Industry-standard platform for design, simulation, and flux estimation in ¹³C-MFA. | Metabolic Flux Analysis Group |
| UniProtKB | Bioinformatics | Provides comprehensive protein sequence annotation, including manually assigned EC numbers. | UniProt Consortium |
The Enzyme Commission (EC) number hierarchical classification system, established in 1961 by the International Union of Biochemistry and Molecular Biology (IUBMB), has been the cornerstone of enzyme nomenclature. This system classifies enzymes into seven main classes based on the chemical reaction they catalyze, using a four-component number (e.g., EC 1.1.1.1 for alcohol dehydrogenase). However, the exponential growth of genomic and metagenomic data, coupled with the discovery of multifunctional and promiscuous enzymes, has exposed significant limitations in the manual, reaction-centric EC framework.
This whitepaper posits that the future of robust, scalable, and accurate enzyme annotation lies in the integration of machine learning (ML) with unified, data-driven frameworks that extend beyond the traditional EC hierarchy.
Quantitative Analysis of the Current EC Landscape
Table 1: Growth of Enzyme Data vs. EC Annotation Completeness
| Metric | 2015 | 2020 | 2024 (Current Estimate) | Source |
|---|---|---|---|---|
| UniProtKB/Swiss-Prot manually annotated entries | ~550,000 | ~570,000 | ~590,000 | UniProt Statistics |
| Total protein sequences in public databases | ~90 million | ~250 million | ~500 million | NCBI, EBI |
| Percentage with EC annotation | ~24% | ~12% | ~5-7% | Derived from UniProt & MGnify |
| Novel EC numbers assigned annually | ~200 | ~150 | ~100 | IUBMB Enzyme Nomenclature |
| Characterized enzymes without EC numbers | N/A | Significant Gap | Estimated 30-40% of literature | Text mining studies |
Table 2: Performance of ML Models for EC Number Prediction
| Model / Tool | Data Source | Prediction Depth | Reported Accuracy (Top-1) | Key Limitation |
|---|---|---|---|---|
| DeepEC (2019) | Protein Sequence | Full 4-level | 91.2% (1st level) | Struggles with remote homology |
| CLEAN (2022) | Enzyme Function (EC) | Enzyme Similarity | 0.973 AUC | Requires known EC similarity |
| ECPred (2021) | Sequence & Structure | Full 4-level | 88.7% (weighted F1) | Dependency on structural data |
| ProtBERT / ESM-2 Fine-Tuning | Language Model Embeddings | 1st & 2nd level | ~94% (1st level) | Computationally intensive; black-box |
Core Methodologies: Experimental & Computational Protocols
Protocol for Generating Training Data for ML Models
Objective: To create a high-quality, non-redundant dataset of enzyme sequences with validated EC numbers.
- Source Data Extraction: Download all reviewed entries from UniProtKB/Swiss-Prot with EC numbers.
- Sequence Clustering: Use CD-HIT at 40% sequence identity threshold to remove redundancy and avoid model bias.
- Data Partitioning: Split the clustered dataset into training (70%), validation (15%), and test (15%) sets, ensuring no two sequences in different sets share >40% identity.
- Feature Engineering: Generate multiple feature sets:
- Sequence Features: Amino acid composition, dipeptide composition, PSSM profiles via PSI-BLAST.
- Evolutionary Features: Pre-computed embeddings from protein language models (e.g., ESM-2, ProtBERT).
- Structure-Based Features: (If available) Secondary structure predictions, solvent accessibility.
- Label Encoding: Format EC numbers as a hierarchical multi-label target (e.g., EC 1.2.3.4 as [1, 2, 3, 4]).
Protocol for a Hybrid Deep Learning Model Architecture
Objective: Implement a hierarchical multi-task learning model that respects the EC tree structure.
- Input Layer: Accepts either raw sequence (one-hot encoded) or a 1280-dimensional protein language model embedding.
- Feature Extraction Backbone: A convolutional neural network (CNN) for sequence patterns or a dense network for embeddings.
- CNN layers: 3 layers with filters 256, 512, 1024, kernel size 5, ReLU activation.
- Hierarchical Prediction Heads:
- Branch 1 (EC First Digit): Dense layer (512 units) → Softmax output (7 classes).
- Branch 2 (EC Second Digit): Concatenate backbone features and first-digit prediction. Dense layer (512 units) → Softmax output (varies by parent class).
- Branches 3 & 4: Similar architecture, each consuming features and all previous level predictions.
- Loss Function: Combined weighted loss: L_total = αL1 + βL2 + γL3 + δL4, where L# are cross-entropy losses.
- Training: Use Adam optimizer (lr=0.001), batch size=64, early stopping on validation loss.
Experimental Validation Protocol for Novel Enzyme Function
Objective: Biochemically validate ML-predicted EC numbers for uncharacterized proteins.
- Protein Expression & Purification: Clone gene of interest into pET vector, express in E. coli BL21(DE3), purify via His-tag affinity chromatography.
- Activity Screening: Set up reaction mixtures containing purified enzyme, predicted substrate (from ML model), and necessary cofactors. Use a multi-well plate format.
- Analytical Detection:
- Spectrophotometric Assay: Monitor NAD(P)H oxidation/reduction at 340 nm.
- Chromatographic Assay (HPLC/GC): For non-chromophoric substrates/products.
- Coupled Enzyme Assays: To detect specific product formation.
- Kinetic Characterization: Determine Km and kcat for confirmed substrates.
- Data Submission: Annotate protein in public database and submit proposal for new EC number to IUBMB if function is novel.
Visualization of Frameworks and Workflows
ML-Driven EC Number Prediction Pipeline
Unified Knowledge Framework for Enzyme Data Integration
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Enzyme Function Validation
| Item | Function in Protocol (Section 3.3) | Example Product/Catalog # | Notes |
|---|---|---|---|
| Cloning & Expression | |||
| pET Expression Vectors | High-yield protein expression in E. coli. | Novagen pET-28a(+) | Allows N-/C-terminal His-tag fusion. |
| Competent E. coli Cells | Protein expression host. | NEB BL21(DE3) | Deficient in proteases for stability. |
| Purification | |||
| Ni-NTA Agarose Resin | Immobilized metal affinity chromatography (IMAC) for His-tagged protein purification. | Qiagen 30210 | High binding capacity, suitable for batch/column. |
| Imidazole | Competes with His-tag for nickel binding; used for elution. | Sigma-Aldrich I2399 | Prepare stock solution at 1M, pH 8.0. |
| Activity Assay | |||
| Cofactor Substrates (NAD(P)H) | Essential for oxidoreductase assays; measurable at 340 nm. | Roche 10128023001 | Light-sensitive; prepare fresh daily. |
| Broad-Substrate Library | High-throughput screening of potential enzyme substrates. | BioVision K589-100 | Contains 100+ metabolic intermediates. |
| Analysis | |||
| Size-Exclusion Chromatography (SEC) Column | Final polishing step; removes aggregates and confirms native oligomeric state. | Cytiva Superdex 200 Increase 10/300 GL | Requires HPLC/FPLC system. |
| Stopped-Flow Spectrophotometer | Measures rapid reaction kinetics (ms-s). | Applied Photophysics SX20 | For fast kinetic characterization. |
The future of enzyme nomenclature necessitates a paradigm shift from a purely manual, reaction-based system (EC) to an integrated, machine-learning-augmented framework. This unified system would leverage a central knowledge graph, combining sequence, structure, kinetic, and genomic context data to generate hierarchical, probabilistic annotations. Such a framework will not replace the EC system but will dynamically inform and expand it, enabling accurate, high-throughput annotation for the vast unexplored enzyme universe, thereby accelerating discovery in synthetic biology, metabolic engineering, and drug development.
Conclusion
The EC number system remains an indispensable, function-centric framework for organizing the vast world of enzymology, providing a common language that connects sequence, structure, and biochemical mechanism. While foundational for database interoperability, pathway analysis, and target identification in drug discovery, researchers must be aware of its limitations regarding promiscuous enzymes and evolutionary relationships. The future lies in the strategic integration of EC numbers with modern sequence, structure, and mechanism-based ontologies, enhanced by machine learning, to create a more dynamic and predictive classification ecosystem. This evolution will be crucial for accelerating discovery in areas like microbiome research, enzyme engineering, and the development of next-generation therapeutics.