ELISA Protocol for Beginners: A Step-by-Step Guide from Setup to Data Analysis
This comprehensive guide provides a clear and actionable introduction to Enzyme-Linked Immunosorbent Assay (ELISA) for researchers, scientists, and drug development professionals new to the technique.
ELISA Protocol for Beginners: A Step-by-Step Guide from Setup to Data Analysis
Abstract
This comprehensive guide provides a clear and actionable introduction to Enzyme-Linked Immunosorbent Assay (ELISA) for researchers, scientists, and drug development professionals new to the technique. It covers the fundamental principles, detailed protocols for various assay formats, common troubleshooting and optimization strategies, and best practices for data validation and interpretation. Readers will gain the practical knowledge needed to confidently design, execute, and analyze ELISA experiments for applications in biomedical research, diagnostics, and therapeutic development.
ELISA Fundamentals: Understanding the Core Principles and Assay Formats
What is ELISA? A Definition and Historical Context
The Enzyme-Linked Immunosorbent Assay (ELISA) is a cornerstone quantitative analytical technique in immunochemistry, enabling the detection and quantification of a specific analyte (typically antibodies, antigens, proteins, or glycoproteins) within a complex biological sample. As a plate-based assay, ELISA leverages the specificity of antigen-antibody binding and the sensitivity of enzyme-mediated chromogenic detection. This guide, framed within the context of a thesis on ELISA protocol for beginners, provides a technical foundation, historical perspective, and current protocols for researchers, scientists, and drug development professionals.
Historical Context and Definition
The ELISA was developed independently by two research groups in 1971: Eva Engvall and Peter Perlmann at Stockholm University in Sweden, and Anton Schuurs and Bauke van Weemen in the Netherlands. Their work built upon the earlier radioimmunoassay (RIA) technique, replacing the radioactive label with an enzyme conjugate, thereby eliminating radiation hazards and improving stability.
Definition: ELISA is an immunoassay where an antigen or antibody is immobilized on a solid surface (typically a polystyrene microtiter plate) and complexed with an antibody or antigen that is linked to a reporter enzyme. Detection is achieved by incubating the complex with a substrate that the enzyme converts to a measurable product, most commonly a colorimetric change. The intensity of the signal is proportional to the concentration of the target analyte in the sample.
Core Principles and Quantitative Data
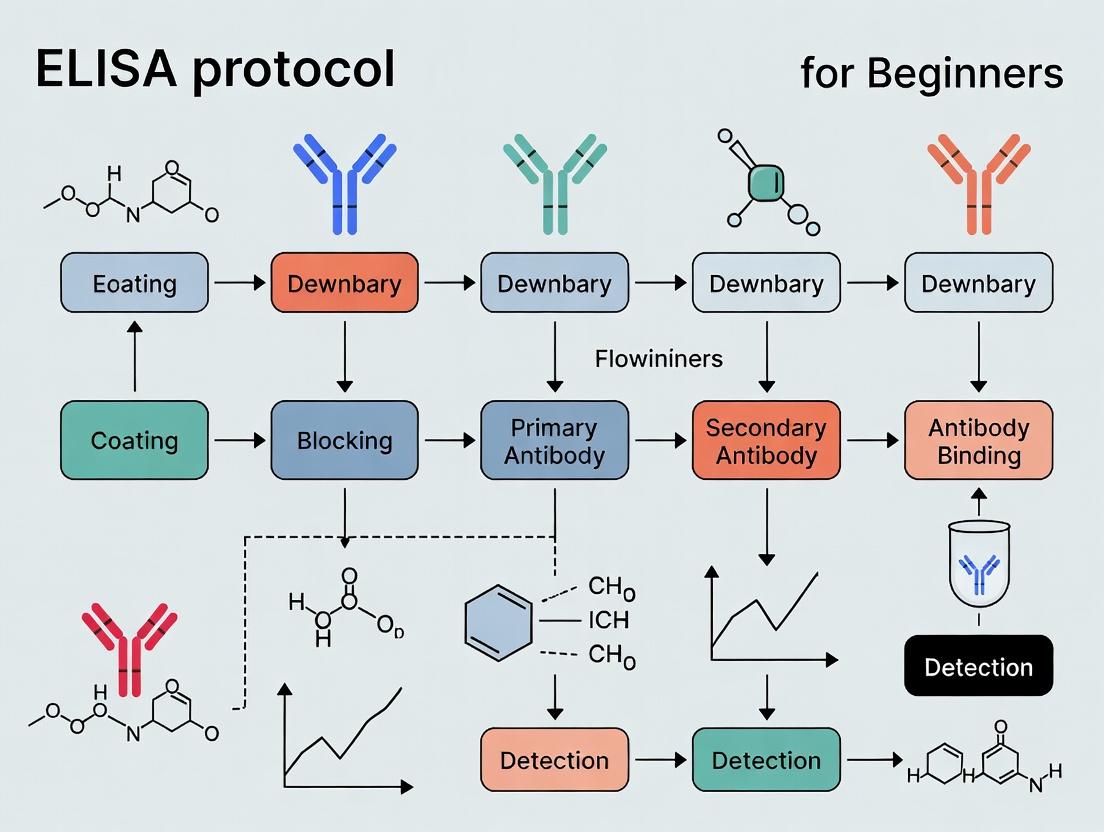
The fundamental principle of ELISA is the specific and high-affinity binding between an antibody and its target antigen. This interaction is then quantified using an enzymatic reaction that amplifies the signal. The general workflow involves: Coating → Blocking → Sample and Detection Antibody Incubation → Substrate Addition → Signal Measurement.
Table 1 summarizes the four main ELISA formats, their primary applications, and relative sensitivities.
Table 1: Core ELISA Formats and Characteristics
| Format | Target Analyte | Immobilized Phase | Detection Conjugate | Key Application | Approx. Sensitivity Range |
|---|---|---|---|---|---|
| Direct ELISA | Antigen | Antigen | Enzyme-linked primary antibody | High-throughput antigen screening | Moderate (ng/mL) |
| Indirect ELISA | Antibody | Antigen | Enzyme-linked secondary antibody | Serology, antibody titer determination | High (pg/mL - ng/mL) |
| Sandwich ELISA | Antigen | Capture antibody | Enzyme-linked detection antibody | Cytokine measurement, biomarker quantification | Very High (pg/mL) |
| Competitive/Inhibition ELISA | Small molecules, antigens | Antigen (or antibody) | Enzyme-linked antigen (or antibody) | Measurement of haptens, cross-reactive antigens | High (pg/mL - ng/mL) |
Detailed Methodology: Sandwich ELISA Protocol
The sandwich ELISA is the most common format for quantifying specific proteins due to its high specificity and sensitivity. The following is a detailed step-by-step protocol suitable for a beginner's research thesis.
Principle: The target antigen is "sandwiched" between a capture antibody bound to the plate and an enzyme-linked detection antibody.
Materials & Reagents (The Scientist's Toolkit): Table 2: Essential Research Reagent Solutions for a Sandwich ELISA
| Item | Function | Typical Example/Concentration |
|---|---|---|
| Coating Buffer | Immobilizes capture antibody on plate via passive adsorption. | Carbonate-bicarbonate buffer, pH 9.6 |
| Wash Buffer | Removes unbound reagents, reducing background. | PBS or Tris buffer with 0.05% Tween 20 (PBST) |
| Blocking Buffer | Covers unsaturated binding sites to prevent nonspecific adsorption. | PBS with 1-5% BSA or 5% non-fat dry milk |
| Capture Antibody | Binds specifically to the target analyte; is immobilized. | Monoclonal antibody, 1-10 µg/mL in coating buffer |
| Detection Antibody | Binds a different epitope on the target analyte; is enzyme-conjugated. | HRP- or AP-linked monoclonal/polyclonal antibody |
| Antigen Standard | Provides known concentrations for generating a standard curve. | Recombinant protein in serial dilutions |
| Enzyme Substrate | Converted by the enzyme to a detectable (e.g., colored) product. | TMB (for HRP), pNPP (for AP) |
| Stop Solution | Halts the enzymatic reaction at a defined timepoint. | 1M H2SO4 (for TMB), 3M NaOH (for pNPP) |
| Microplate Reader | Measures absorbance of the developed color. | Spectrophotometer capable of reading 96/384-well plates |
Experimental Protocol:
- Coating: Dilute the capture antibody in coating buffer. Add 100 µL per well to a 96-well microtiter plate. Seal the plate and incubate overnight at 4°C (or 1-2 hours at 37°C).
- Washing: Aspirate the liquid from each well. Wash the plate 3 times with 300 µL of wash buffer per well. Blot the plate on clean paper towels to remove residual liquid.
- Blocking: Add 200-300 µL of blocking buffer to each well. Incubate for 1-2 hours at room temperature (or 37°C). Wash as in Step 2.
- Sample & Standard Addition: Prepare serial dilutions of the antigen standard in the same buffer as the samples (e.g., assay diluent). Add 100 µL of standard, sample, or negative control to appropriate wells. Incubate for 2 hours at room temperature. Wash as in Step 2.
- Detection Antibody Incubation: Add 100 µL of the enzyme-conjugated detection antibody (diluted in blocking buffer) to each well. Incubate for 1-2 hours at room temperature. Wash as in Step 2.
- Substrate Addition: Add 100 µL of freshly prepared enzyme substrate (e.g., TMB) to each well. Incubate in the dark for 15-30 minutes at room temperature. Monitor color development.
- Stop Reaction: Add 100 µL of stop solution (e.g., 1M H2SO4) to each well. The blue color (if using TMB) will turn yellow.
- Measurement & Analysis: Read the absorbance of each well immediately using a microplate reader at the appropriate wavelength (e.g., 450 nm for TMB). Plot the mean absorbance of the standard concentrations against their known values using a 4- or 5-parameter logistic (4PL/5PL) curve fit. Interpolate the concentration of unknown samples from this standard curve.
Visualization: ELISA Workflow and Signaling
This whitepaper, framed within a broader thesis on ELISA protocol for beginners, provides an in-depth technical guide to the four core components of an Enzyme-Linked Immunosorbent Assay (ELISA). Designed for researchers, scientists, and drug development professionals, this document details the function, selection criteria, and experimental integration of the plate, capture and detection antibodies, enzyme, and substrate. Mastery of these components is fundamental to developing robust, sensitive, and quantitative immunoassays for research and diagnostic applications.
The Core Components: Function and Selection
The Microplate: The Solid Phase Foundation
The microtiter plate serves as the solid phase for immobilizing the target molecule. Its surface chemistry is critical for assay performance.
Selection Criteria:
- Binding Capacity: Measured in ng/cm², it determines how much capture protein can be immobilized.
- Material: Polystyrene is standard; polypropylene may be used for special applications.
- Surface Treatment: High-binding plates (e.g., coated with poly-L-lysine or treated with plasma) are used for proteins/antibodies. Medium-binding plates are suitable for sticky proteins (e.g., streptavidin), and low-binding plates prevent non-specific adsorption.
Table 1: Microplate Surface Types and Characteristics
| Surface Type | Typical Binding Capacity (IgG) | Common Coating/Chemistry | Best For |
|---|---|---|---|
| High-Binding | 300-500 ng/cm² | Passive hydrophobic adsorption, Poly-L-lysine | Capture antibodies, antigens in indirect/direct ELISA |
| Medium-Binding | 100-300 ng/cm² | Mildly hydrophilic | Avidin/Biotin systems, sticky proteins |
| Low-Binding | < 100 ng/cm² | Hydrophilic polymers, Non-fouling surfaces | Sample dilution, reagent storage |
Capture and Detection Antibodies: The Specificity Core
The antibody pair forms the heart of a sandwich ELISA, providing specificity.
- Capture Antibody: Immobilized on the plate, it specifically binds the target antigen from the sample.
- Detection Antibody: Binds to a different epitope on the captured antigen, creating the "sandwich." It is conjugated to an enzyme for signal generation.
Critical Parameters:
- Specificity & Affinity: Must be high to minimize cross-reactivity and ensure efficient capture/detection.
- Epitope Recognition: The pair must recognize non-overlapping epitopes to avoid steric hindrance.
- Host Species: Must be different to prevent interference from secondary antibodies if used.
Table 2: Antibody Conjugation Formats in ELISA
| Format | Detection Antibody Conjugate | Typical Sensitivity Range | Key Advantage |
|---|---|---|---|
| Direct | Enzyme (e.g., HRP) | Moderate | Fast, fewer steps, minimal background |
| Indirect | Biotin or unlabeled | High | Signal amplification via enzyme-streptavidin or secondary Ab |
| Competitive | Enzyme-labeled antigen | Variable (for small molecules) | Ideal for haptens/small antigens with single epitope |
The Enzyme: The Signal Generator
The enzyme conjugated to the detection system catalyzes the conversion of a substrate into a detectable product. Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP) are most common.
Table 3: Common Enzyme Conjugates in ELISA
| Enzyme | Common Source | Optimal pH | Typical Substrate | Signal Readout |
|---|---|---|---|---|
| Horseradish Peroxidase (HRP) | Armoracia rusticana | 5.0 - 7.0 | TMB, OPD, ABTS | Colorimetric (450nm, 492nm, 405nm), Chemiluminescent |
| Alkaline Phosphatase (AP) | Calf intestinal | 9.0 - 10.0 | pNPP, BCIP/NBT | Colorimetric (405nm), Chemifluorescent |
| β-Galactosidase | E. coli | 6.0 - 8.0 | ONPG, MUG | Colorimetric (420nm), Fluorescent |
The Substrate: The Measurable Output
The substrate choice dictates the signal type (colorimetric, chemiluminescent, fluorescent) and assay sensitivity.
Table 4: Common ELISA Substrates and Their Properties
| Substrate | For Enzyme | Product Type | Readout Method | Sensitivity (approx.) |
|---|---|---|---|---|
| 3,3',5,5'-Tetramethylbenzidine (TMB) | HRP | Soluble Blue (Yellow after acid stop) | Colorimetric (450nm) | 10-100 pg/mL |
| p-Nitrophenyl Phosphate (pNPP) | AP | Soluble Yellow | Colorimetric (405nm) | 1-10 ng/mL |
| Enhanced Chemiluminescent (ECL) | HRP | Light | Luminometer | 0.1-1 pg/mL |
| 4-Methylumbelliferyl phosphate (4-MUP) | AP | Fluorescent | Fluorometer (Ex 360nm/Em 440nm) | 1-10 pg/mL |
Experimental Protocol: Standard Sandwich ELISA Workflow
Title: Coating the plate with capture antibody. Protocol 1: Plate Coating
- Dilute the capture antibody in a suitable carbonate/bicarbonate buffer (pH 9.6) or PBS (pH 7.4) to a concentration typically between 1-10 µg/mL.
- Dispense 50-100 µL per well into a high-binding polystyrene microplate.
- Seal the plate and incubate overnight at 4°C (or 1-2 hours at 37°C).
- Aspirate the coating solution and wash the plate 3 times with 200-300 µL of wash buffer (PBS or Tris with 0.05% Tween 20, PBST/TBST). Blot dry.
Title: Blocking to prevent non-specific binding. Protocol 2: Blocking
- Add 200-300 µL of blocking buffer (e.g., 1-5% BSA or non-fat dry milk in PBST) to each well.
- Incubate at room temperature for 1-2 hours (or 37°C for 1 hour).
- Aspirate and wash as in Protocol 1, step 4. The plate is now ready for sample addition.
Title: Antigen capture and detection. Protocol 3: Antigen Incubation & Detection
- Add 50-100 µL of sample or antigen standard (diluted in blocking or assay buffer) to appropriate wells. Include blanks (buffer only). Incubate 1-2 hours at RT or 37°C. Wash.
- Add 50-100 µL of the detection antibody (diluted per manufacturer's recommendation in blocking buffer). Incubate 1-2 hours at RT. Wash.
- (If using an indirect/biotin system): Add enzyme-conjugated Streptavidin or secondary antibody. Incubate 30 mins - 1 hour at RT. Wash.
- Prepare the chosen substrate solution immediately before use. Add 50-100 µL per well.
- Incubate in the dark for a defined period (e.g., 5-30 minutes for TMB).
- If required, add an equal volume of stop solution (e.g., 1M H₂SO₄ for TMB).
- Read absorbance immediately on a plate reader at the appropriate wavelength.
Visualizing the ELISA Workflow and Signaling
Diagram Title: Step-by-step workflow of a sandwich ELISA protocol.
Diagram Title: Molecular architecture and signal generation in sandwich ELISA.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 5: Key Reagents and Materials for ELISA Development
| Item | Function & Description | Typical Example/Note |
|---|---|---|
| High-Binding Polystyrene Plate | Solid phase for protein adsorption. | Corning Costar 9018, Nunc MaxiSorp. |
| Coating Buffer (pH 9.6) | Optimizes passive adsorption of capture antibody. | 0.1 M Carbonate/Bicarbonate buffer. |
| Blocking Buffer | Saturates unused protein-binding sites to reduce background. | 1-5% BSA in PBST, or proprietary protein-free blockers. |
| Wash Buffer | Removes unbound material; detergents reduce non-specific binding. | PBS or Tris + 0.05% - 0.1% Tween 20 (PBST/TBST). |
| Matched Antibody Pair | Monoclonal or affinity-purified polyclonal antibodies for capture/detection. | Must bind non-overlapping epitopes. Validated pairs are recommended. |
| Detection Antibody Conjugate | Antibody linked directly or indirectly to signal-generating enzyme. | HRP-conjugated detection Ab or biotinylated Ab + Streptavidin-HRP. |
| Chromogenic Substrate | Enzyme substrate producing a colored, measurable product. | TMB (colorimetric, HRP), pNPP (colorimetric, AP). |
| Stop Solution | Halts the enzyme-substrate reaction at a defined timepoint. | 1M H₂SO₄ (for TMB), 3M NaOH (for pNPP). |
| Plate Reader | Instrument to quantify colorimetric, fluorescent, or luminescent signal. | Filter-based or monochromator-based multimode readers. |
Within the context of a broader thesis on ELISA protocol for beginners research, this whitepaper serves as an in-depth technical guide to the four core assay formats. Enzyme-Linked Immunosorbent Assay (ELISA) remains a cornerstone technique in life sciences, clinical diagnostics, and drug development for the detection and quantification of proteins, peptides, antibodies, and hormones. Selecting the appropriate format is fundamental to experimental success, balancing factors such as sensitivity, specificity, required reagents, and procedural complexity. This guide details the principles, methodologies, and applications of Direct, Indirect, Sandwich, and Competitive ELISAs to empower researchers in making informed protocol decisions.
Direct ELISA
Principle: The target antigen is immobilized directly onto the surface of a polystyrene microplate well. A primary antibody conjugated to an enzyme (e.g., Horseradish Peroxidase, HRP) is then added, which binds specifically to the antigen. After washing, a chromogenic substrate is added, and the enzyme catalyzes a reaction producing a measurable color signal proportional to the amount of antigen present.
Advantages and Disadvantages:
- Pros: Simple and rapid protocol with minimal steps; eliminates cross-reactivity from secondary antibodies.
- Cons: Lower sensitivity due to fewer signal amplification steps; requires labeling every primary antibody, which can be costly and time-consuming.
Detailed Protocol for Direct ELISA:
- Coating: Dilute the antigen in carbonate-bicarbonate coating buffer (pH 9.6) to 1-10 µg/mL. Add 100 µL per well of a 96-well microplate. Seal and incubate overnight at 4°C.
- Washing: Aspirate the coating solution. Wash the plate 3 times with 300 µL of Wash Buffer (e.g., PBS with 0.05% Tween-20, PBST). Blot plate on absorbent paper.
- Blocking: Add 200-300 µL of Blocking Buffer (e.g., 1-5% BSA or non-fat dry milk in PBST) per well. Incubate for 1-2 hours at room temperature (RT). Wash as in Step 2.
- Primary Antibody Incubation: Add 100 µL of the enzyme-conjugated primary antibody, diluted in Blocking Buffer, to each well. Incubate for 1-2 hours at RT. Wash thoroughly (3-5 times).
- Detection: Prepare the enzyme substrate solution (e.g., TMB for HRP). Add 100 µL per well. Incubate in the dark for 5-30 minutes at RT until color develops.
- Stop & Read: Add 50-100 µL of Stop Solution (e.g., 1M H₂SO₄ for TMB) to each well. Measure absorbance immediately at the appropriate wavelength (e.g., 450nm for TMB) using a microplate reader.
Title: Direct ELISA Workflow
Indirect ELISA
Principle: The antigen is immobilized on the plate. An unlabeled primary antibody binds to the antigen. Subsequently, an enzyme-conjugated secondary antibody, which recognizes the Fc region of the host species of the primary antibody, is added. This two-step process provides signal amplification.
Advantages and Disadvantages:
- Pros: High sensitivity due to amplification from multiple secondary antibodies binding to a single primary; great flexibility as one labeled secondary antibody can be used with many primary antibodies from the same host species.
- Cons: Increased time and steps; potential for cross-reactivity or higher background if secondary antibody is not specific.
Detailed Protocol for Indirect ELISA:
- Coating & Blocking: Perform as described in the Direct ELISA protocol (Steps 1-3).
- Primary Antibody Incubation: Add 100 µL of unlabeled primary antibody, diluted in Blocking Buffer, to each well. Incubate for 1-2 hours at RT (or overnight at 4°C for higher sensitivity). Wash 3-5 times.
- Secondary Antibody Incubation: Add 100 µL of enzyme-conjugated secondary antibody (e.g., Goat anti-Mouse HRP), diluted in Blocking Buffer, to each well. Incubate for 1-2 hours at RT in the dark. Wash 5-7 times thoroughly.
- Detection & Stop: Perform as described in the Direct ELISA protocol (Steps 5-6).
Title: Indirect ELISA Workflow
Sandwich ELISA
Principle: The most common format for detecting antigens, especially complex samples. A capture antibody is immobilized on the plate. The sample containing the antigen is added, and the antigen is "captured." A second, detector antibody (often enzyme-conjugated or biotinylated) binds to a different epitope on the antigen, forming an antibody-antigen-antibody "sandwich."
Advantages and Disadvantages:
- Pros: Very high specificity and sensitivity; ideal for complex samples (e.g., serum, cell lysates) as the antigen does not need to be purified before the assay.
- Cons: Requires two antibodies that bind to non-overlapping epitopes on the target antigen; more optimization is needed.
Detailed Protocol for Sandwich ELISA:
- Capture Antibody Coating: Dilute the capture antibody in coating buffer to 2-10 µg/mL. Add 100 µL per well. Seal and incubate overnight at 4°C.
- Washing & Blocking: Aspirate and wash 3 times. Add Blocking Buffer and incubate for 1-2 hours at RT. Wash.
- Antigen Incubation: Add 100 µL of sample or antigen standard (diluted in Blocking Buffer) to each well. Incubate for 2 hours at RT (or overnight at 4°C). Wash 3-5 times.
- Detection Antibody Incubation: Add 100 µL of the enzyme-conjugated detection antibody, diluted in Blocking Buffer. Incubate for 1-2 hours at RT in the dark. Wash 5-7 times. (Note: For indirect detection, use an unlabeled detection antibody followed by an enzyme-conjugated tertiary antibody).
- Detection & Stop: Perform as described in the Direct ELISA protocol (Steps 5-6).
Title: Sandwich ELISA Workflow
Competitive ELISA
Principle: Used to measure small antigens or antigens with only a single antibody-binding site. The target antigen in the sample competes with a labeled reference antigen for a limited number of antibody-binding sites immobilized on the plate. The signal is inversely proportional to the amount of target antigen in the sample.
Advantages and Disadvantages:
- Pros: Highly specific for small molecules; can be used with complex samples; less susceptible to sample matrix effects.
- Cons: More complex data analysis; requires a purified, labeled competitor antigen.
Detailed Protocol for Competitive ELISA (Antigen-Coated Format):
- Coating & Blocking: Coat the plate with purified antigen (2-5 µg/mL). Incubate overnight at 4°C. Wash and block as previously described.
- Competition Reaction: Pre-mix a constant, limiting amount of primary antibody with serially diluted samples or standards containing the unknown antigen. Incubate this mixture for 1-2 hours at RT to allow competition.
- Transfer & Incubation: Transfer the antibody-sample/standard mixtures to the antigen-coated plate. Incubate for 30-60 minutes at RT. The free antibody will bind to the immobilized antigen. Wash 3-5 times.
- Secondary Antibody & Detection: Add enzyme-conjugated secondary antibody (if the primary is unlabeled). Incubate, wash, and proceed with detection as in an Indirect ELISA. Higher sample antigen concentration yields lower final signal.
Title: Competitive ELISA Principle: Antigen Competition
Quantitative Comparison of ELISA Formats
Table 1: Key Characteristics of the Four Main ELISA Formats
| Format | Sensitivity | Specificity | Complexity | Time Required | Key Applications |
|---|---|---|---|---|---|
| Direct | Low (ng/mL) | Moderate | Low | ~4-5 hours | Quick screening of high-abundance targets; antibody epitope mapping. |
| Indirect | High (pg/mL - ng/mL) | High | Moderate | ~5-6 hours | Serum antibody detection (e.g., immunogenicity, infectious disease). |
| Sandwich | Very High (pg/mL) | Very High | High | ~6-8 hours (or overnight) | Quantifying cytokines, biomarkers, hormones in complex biological fluids. |
| Competitive | Moderate to High (pg/mL - ng/mL) | Very High | High | ~5-7 hours | Measuring small molecules (haptens): drugs, hormones (estradiol, T3/T4). |
Table 2: Typical Reagent Concentrations and Incubation Times for ELISA Protocols
| Step | Direct ELISA | Indirect ELISA | Sandwich ELISA | Competitive ELISA |
|---|---|---|---|---|
| Coating | Antigen: 1-10 µg/mL, O/N @ 4°C | Antigen: 1-10 µg/mL, O/N @ 4°C | Capture Ab: 2-10 µg/mL, O/N @ 4°C | Antigen: 2-5 µg/mL, O/N @ 4°C |
| Blocking | 1-5% protein, 1-2h @ RT | 1-5% protein, 1-2h @ RT | 1-5% protein, 1-2h @ RT | 1-5% protein, 1-2h @ RT |
| Primary Ab | Conjugated: 0.5-2 µg/mL, 1-2h @ RT | Unlabeled: 0.1-2 µg/mL, 1-2h @ RT | Sample/Antigen: Variable, 2h @ RT | Pre-mix step: Antibody + Sample, 1-2h @ RT |
| Secondary Ab | Not Required | Conjugated: 0.5-2 µg/mL, 1-2h @ RT | Detector Ab (Conj.): 0.5-2 µg/mL, 1-2h @ RT | Conjugated: 0.5-2 µg/mL, 1-2h @ RT |
| Detection | Substrate: 5-30 min @ RT, protected from light. | Substrate: 5-30 min @ RT, protected from light. | Substrate: 5-30 min @ RT, protected from light. | Substrate: 5-30 min @ RT, protected from light. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for ELISA Experiments
| Item | Function | Key Considerations |
|---|---|---|
| Polystyrene Microplates | Solid phase for immobilizing biomolecules. | High-binding plates (e.g., Nunc MaxiSorp) for proteins/antibodies; medium-binding for other molecules. |
| Coating Buffer (Carbonate-Bicarbonate, pH 9.6) | Optimal pH for passive adsorption of proteins to polystyrene. | pH is critical for efficient binding. Filter before use. |
| Wash Buffer (PBS with 0.05% Tween-20) | Removes unbound reagents, reduces non-specific background. | Tween-20 concentration can be optimized; ensure consistent washing. |
| Blocking Buffer (e.g., BSA, Casein, Non-fat Dry Milk) | Covers uncovered plastic surface to prevent non-specific binding of detection antibodies. | Must be compatible with all assay components. BSA is standard; casein offers low background for phospho-targets. |
| Primary Antibodies | Specifically bind the target analyte. | Validate for application (ELISA). Check host species, clonality, and required concentration. |
| Enzyme-Conjugated Secondary Antibodies | Bind to primary antibodies for signal generation (Indirect/Sandwich). | Must be raised against the host species of the primary antibody. HRP and Alkaline Phosphatase (AP) are common. |
| Chromogenic Substrates (TMB, OPD, pNPP) | Converted by enzyme to a colored, measurable product. | TMB (HRP) is most common, yielding blue color (450nm read) stopped to yellow by acid. Choose based on enzyme and required sensitivity. |
| Stop Solution (e.g., 1M H₂SO₄, 2M NaOH) | Halts the enzymatic reaction, stabilizing signal for reading. | Must match the substrate (acid stop for TMB/OPD, base for pNPP). |
| Microplate Reader | Measures absorbance (optical density, OD) of the stopped reaction. | Must have appropriate filters (e.g., 450nm for acid-stopped TMB). |
Mastery of the four main ELISA formats—Direct, Indirect, Sandwich, and Competitive—provides a foundational toolkit for quantitative protein analysis. For beginners, the Indirect ELISA often offers an excellent balance of sensitivity and reagent flexibility. For complex sample analysis, the Sandwich ELISA is the gold standard, while the Competitive format is indispensable for small molecule quantification. Success hinges on careful reagent selection, meticulous protocol optimization—particularly of antibody pairings in Sandwich ELISA—and robust data analysis. By understanding the principles and trade-offs outlined in this guide, researchers can strategically select and execute the optimal ELISA protocol to generate reliable, publication-quality data.
1. Introduction
Within the foundational thesis of "ELISA Protocol for Beginners Research," selecting the appropriate assay format is the first and most critical decision. The Enzyme-Linked Immunosorbent Assay (ELISA) is not a single method but a family of formats, each with distinct architectures for antibody-antigen interaction. This guide provides an in-depth technical comparison of core ELISA formats, detailing their applications, experimental protocols, and inherent limitations to inform robust experimental design for researchers, scientists, and drug development professionals.
2. Core ELISA Formats: A Comparative Analysis
The choice of format dictates sensitivity, specificity, required reagents, and time. The four primary formats are summarized below.
Table 1: Comparative Overview of Core ELISA Formats
| Format | Principle | Key Advantages | Key Limitations | Primary Applications |
|---|---|---|---|---|
| Direct ELISA | Antigen is immobilized and detected directly by a labeled primary antibody. | • Fastest procedure (fewer steps).• Minimal cross-reactivity from secondary antibodies. | • Low signal amplification (low sensitivity).• Requires labeling of every primary antibody.• Potential for high background. | • Screening antibody-antigen binding.• Antigen detection when high specificity is assured. |
| Indirect ELISA | Immobilized antigen is detected by an unlabeled primary antibody, which is then bound by a labeled secondary antibody. | • High sensitivity due to signal amplification (multiple secondary Abs bind per primary).• Flexible–one labeled secondary antibody can be used for many primary antibodies.• Maximum immunoreactivity of primary antibody is maintained. | • Longer protocol.• Risk of cross-reactivity from secondary antibody.• Requires species-specific secondary antibodies. | • Most common format for total antibody detection (e.g., serology).• General purpose antigen detection. |
| Sandwich ELISA | Capture antibody immobilizes antigen, which is then detected by a second, labeled detection antibody. | • High specificity (two antibodies required).• High sensitivity and precision.• Suitable for complex samples (antigen does not need purification). | • Requires two antibodies that bind different, non-overlapping epitopes.• More optimization required (antibody pairing).• Not suitable for small antigens/haptens (<~500 Da). | • Quantification of specific proteins in complex mixtures (e.g., cytokines, biomarkers).• Clinical diagnostics. |
| Competitive/Inhibition ELISA | Sample antigen competes with a reference antigen for binding to a limited amount of labeled antibody. Signal is inversely proportional to analyte concentration. | • Can measure small antigens/haptens.• Tolerates complex sample matrices.• High specificity when purified antigen is available. | • More complex data interpretation.• Narrower dynamic range.• Lower overall sensitivity compared to sandwich. | • Measurement of small molecules (hormones, drugs).• Detection of antigens with only one available antibody epitope. |
3. Detailed Experimental Protocols
Protocol A: Indirect ELISA for Antibody Titration (from Serum)
- Day 1: Coating
- Dilute purified antigen in carbonate-bicarbonate coating buffer (pH 9.6) to 1-10 µg/mL.
- Add 100 µL per well to a 96-well microplate. Seal and incubate overnight at 4°C.
- Day 2: Blocking, Primary & Secondary Antibody Incubation
- Discard coating solution. Wash plate 3x with 300 µL PBS-T (0.05% Tween-20) per well.
- Add 300 µL blocking buffer (5% non-fat dry milk or BSA in PBS) per well. Incubate 1-2 hours at room temperature (RT).
- Wash plate 3x with PBS-T.
- Prepare serial dilutions of test serum in blocking buffer. Add 100 µL per well. Incubate 1-2 hours at RT.
- Wash plate 3x with PBS-T.
- Dilute enzyme-conjugated secondary antibody (e.g., HRP-anti-species IgG) in blocking buffer per manufacturer’s instructions. Add 100 µL per well. Incubate 1-2 hours at RT in the dark.
- Wash plate 3x with PBS-T.
- Day 2: Detection & Analysis
- Prepare TMB substrate solution. Add 100 µL per well. Incubate in the dark for 5-30 minutes until blue color develops.
- Stop reaction by adding 50 µL of 2N H₂SO₄ (color changes to yellow).
- Immediately read absorbance at 450 nm using a plate reader.
Protocol B: Sandwich ELISA for Cytokine Quantification
- Day 1: Capture Antibody Coating
- Dilute capture antibody in PBS (pH 7.4) to 2-10 µg/mL.
- Add 100 µL per well. Seal and incubate overnight at 4°C.
- Day 2: Blocking, Sample & Detection Antibody Incubation
- Wash plate 3x with PBS-T.
- Block with 300 µL blocking buffer (1% BSA in PBS) for 1-2 hours at RT.
- Wash plate 3x with PBS-T.
- Add 100 µL of sample or standard (diluted in blocking buffer) per well. Incubate 2 hours at RT or overnight at 4°C.
- Wash plate 3-5x with PBS-T.
- Add 100 µL of biotinylated detection antibody (diluted in blocking buffer) per well. Incubate 1-2 hours at RT.
- Wash plate 3-5x with PBS-T.
- Add 100 µL of Streptavidin-HRP conjugate (diluted per manufacturer) per well. Incubate 30 minutes at RT in the dark.
- Wash plate 3-5x with PBS-T.
- Day 2: Detection & Analysis
- Add 100 µL TMB substrate. Incubate in the dark for 5-20 minutes.
- Stop with 50 µL 2N H₂SO₄.
- Read absorbance at 450 nm. Generate a standard curve for quantification.
4. Visualization of ELISA Formats and Workflows
Diagram 1: Direct vs Indirect ELISA Workflow
Diagram 2: Sandwich ELISA Antibody-Antigen Binding
5. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for ELISA Development
| Reagent/Material | Function & Critical Considerations |
|---|---|
| Microplate | Solid phase for immobilization. High-binding (e.g., polystyrene) is standard. Chemically modified plates (e.g., streptavidin-coated) enable specific formats. |
| Coating Buffer (Carbonate-Bicarbonate, pH 9.6) | Alkaline buffer promotes passive adsorption of proteins (antigens/antibodies) to the plastic surface via hydrophobic interactions. |
| Blocking Buffer (e.g., BSA, Casein, Non-Fat Dry Milk) | Saturates remaining protein-binding sites on the plate after coating to prevent non-specific binding of detection reagents, reducing background noise. |
| Wash Buffer (PBS with 0.05% Tween-20) | Removes unbound reagents between steps. Tween-20 (a non-ionic detergent) reduces non-specific binding. Consistent washing is critical for precision. |
| Detection Antibodies | Primary antibodies bind the analyte. Conjugated secondary antibodies (e.g., HRP-anti-IgG) enable signal generation. Biotinylated antibodies allow further amplification via streptavidin-enzyme complexes. |
| Enzyme Substrate (e.g., TMB, OPD) | Chromogenic substrate for HRP (Horseradish Peroxidase) or AP (Alkaline Phosphatase) enzymes. Produces a measurable color change. Must be stable and have low background. |
| Stop Solution (e.g., 2N H₂SO₄ for TMB) | Halts the enzyme-substrate reaction at a defined endpoint, stabilizing the final signal for measurement. |
| Reference Standards & Controls | Purified analyte of known concentration for generating a standard curve. Positive and negative controls are mandatory for assay validation and troubleshooting. |
6. Conclusion
The "right" ELISA format is determined by the experimental question, the nature of the analyte (size, availability, abundance), and the reagent landscape. For beginners, the Indirect ELISA offers a robust introduction to core principles. For specific protein quantification, the Sandwich ELISA provides superior specificity and sensitivity, despite increased complexity. Understanding the applications, advantages, and limitations of each format, as outlined in this guide, is fundamental to designing a reliable and reproducible ELISA within any research or diagnostic pipeline.
Essential Reagents and Equipment for a Basic ELISA Setup
Within the broader thesis of establishing a robust ELISA protocol for beginners in research, this guide details the core reagents and equipment required for a basic setup. ELISA (Enzyme-Linked Immunosorbent Assay) is a foundational technique for detecting and quantifying proteins, peptides, antibodies, or hormones. A successful experiment hinges on the quality and proper application of these essential components.
Core Reagents and Their Functions
The following table outlines the essential reagents, their primary function, and key considerations for a typical sandwich ELISA.
Table 1: Essential Reagents for a Sandwich ELISA
| Reagent | Primary Function | Key Considerations |
|---|---|---|
| Solid Phase (Plate) | Provides surface for antigen immobilization. | High-binding 96-well polystyrene plates are standard. Ensure compatibility with your detector. |
| Capture Antibody | Binds specifically to target antigen, immobilizing it on the plate. | Must be specific, high-affinity, and used at optimal coating concentration. |
| Detection Antibody | Binds to a different epitope on the captured antigen. | Conjugated to an enzyme (e.g., HRP). Specificity and affinity are critical. |
| Target Antigen (Standard & Sample) | The molecule of interest to be quantified. | A purified standard of known concentration is mandatory for generating a calibration curve. |
| Blocking Buffer | Covers unused protein-binding sites to reduce non-specific background. | Typically 1-5% BSA or non-fat dry milk in a compatible buffer (e.g., PBS). |
| Wash Buffer | Removes unbound reagents between steps to minimize background. | Usually PBS or Tris buffer with a detergent (e.g., 0.05% Tween 20). |
| Enzyme Substrate | Converted by the conjugated enzyme to a detectable (chromogenic/fluorogenic) product. | Common: TMB (colorimetric, read at 450nm). Must match the enzyme (HRP uses TMB/OPD). |
| Stop Solution | Halts the enzyme-substrate reaction at a defined time. | For TMB: Typically a strong acid (e.g., 1M H₂SO₄ or HCl), changing color from blue to yellow. |
| Coating Buffer | Provides optimal pH and ionic strength for passive adsorption of the capture antibody. | Commonly carbonate/bicarbonate buffer, pH 9.6. |
Essential Equipment and Instrumentation
Table 2: Essential Equipment for ELISA Execution
| Equipment | Primary Function | Specification Notes |
|---|---|---|
| Microplate Reader | Measures the absorbance (or fluorescence/chemiluminescence) of each well. | Filter-based or monochromator-based. Must match the substrate's detection mode (e.g., 450nm for TMB). |
| Microplate Washer (or Bottle/Manifold) | Automated or manual washing of plate wells to remove unbound material. | Automated washers improve reproducibility. Manual washing requires a multichannel pipette and a wash bottle. |
| Single & Multichannel Pipettes | Accurate and precise liquid handling for reagents and samples. | Critical for reproducibility. Typical volumes: 50-100 µL for reagents, 100-200 µL for wash steps. |
| Incubator/Shaker | Maintains consistent temperature and gentle agitation during incubations. | Typically 37°C for shorter incubations; 4°C for overnight coating. Shaking improves binding kinetics. |
| Plate Sealer | Covers plates during incubations to prevent evaporation and contamination. | Adhesive film or reusable plate lids. |
| Data Analysis Software | Analyzes raw data, generates standard curves, and calculates sample concentrations. | Built into plate readers or standalone (e.g., Prism, Excel with curve-fitting). |
Detailed Protocol for a Direct Sandwich ELISA
Methodology:
- Coating: Dilute the capture antibody in coating buffer. Dispense 100 µL per well into a 96-well microplate. Seal and incubate overnight at 4°C or for 1-2 hours at room temperature (RT).
- Washing: Aspirate liquid from wells. Wash each well 3 times with 200-300 µL of wash buffer, ensuring complete removal of liquid between washes.
- Blocking: Add 200-300 µL of blocking buffer to each well. Incubate for 1-2 hours at RT or 4°C overnight. Wash as in step 2.
- Antigen Incubation: Prepare serial dilutions of the standard antigen in the sample diluent/buffer. Add 100 µL of standards and prepared samples to appropriate wells. Incubate for 2 hours at RT or 1 hour at 37°C. Wash 3 times.
- Detection Antibody Incubation: Add 100 µL of the enzyme-conjugated detection antibody (diluted in blocking buffer) to each well. Incubate for 1-2 hours at RT. Wash 3-5 times thoroughly.
- Substrate Addition: Add 100 µL of freshly prepared substrate solution (e.g., TMB) to each well. Incubate in the dark for 10-30 minutes at RT.
- Stop Reaction: Add 50-100 µL of stop solution to each well. The color change indicates reaction cessation.
- Measurement: Read the absorbance of each well at the appropriate wavelength (e.g., 450nm for acid-stopped TMB) within 30 minutes.
Diagram Title: Basic Sandwich ELISA Workflow
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions
| Item | Function in ELISA |
|---|---|
| High-Binding Polystyrene Plate | The solid phase platform that passively adsorbs proteins via hydrophobic interactions. |
| Phosphate-Buffered Saline (PBS) with Tween-20 | The basis for wash buffer; the ionic strength cleans wells, while the detergent reduces non-specific binding. |
| Bovine Serum Albumin (BSA) Blocking Solution | A standard blocking agent that saturates empty protein-binding sites on the plate to prevent false-positive signals. |
| TMB (3,3',5,5'-Tetramethylbenzidine) Substrate | A chromogenic substrate for Horseradish Peroxidase (HRP). Yields a blue product measurable at 650nm or, after acid stop, yellow at 450nm. |
| Horseradish Peroxidase (HRP) Conjugate | The enzyme commonly linked to the detection antibody; catalyzes the conversion of the substrate to a detectable signal. |
Diagram Title: Sandwich ELISA Molecular Recognition
A Step-by-Step ELISA Protocol: From Plate Coating to Signal Detection
Within the broader thesis on mastering the ELISA protocol for beginners, Step 2—incubation of the sample and subsequent detection antibodies—is the critical juncture where assay specificity and sensitivity are determined. This phase involves the precise binding of target antigens by capture antibodies and their detection by enzyme-conjugated antibodies. Optimization of concentrations and incubation times here is paramount to reduce non-specific binding, minimize background noise, and maximize signal-to-noise ratios, forming the cornerstone of reliable, reproducible data for researchers, scientists, and drug development professionals.
Foundational Principles of Incubation Optimization
The incubation process is governed by kinetic principles. Antigen-antibody binding is not instantaneous; it requires time to reach equilibrium. The primary goals are to ensure sufficient time for complete binding while maintaining a practical workflow. Key variables are:
- Antibody Concentration: Too high increases cost and background; too low reduces signal and assay dynamic range.
- Incubation Time: Longer times can increase binding but also prolong total assay time and potentially increase non-specific interactions.
- Temperature: Most assays use 37°C for faster kinetics or 4°C overnight for high-affinity binding with lower background.
- Sample Composition: Matrix effects (serum, plasma, cell lysate) can interfere, necessitating optimization in the relevant buffer.
Quantitative Optimization Data
Recent literature and vendor application notes provide a framework for optimization ranges. The following tables summarize key quantitative data for standard sandwich ELISA formats.
Table 1: Optimization Ranges for Capture Antibody Coating (Step 1, for context)
| Parameter | Typical Range | Recommended Starting Point | Comments |
|---|---|---|---|
| Concentration | 1–10 µg/mL in PBS | 2–5 µg/mL | High-affinity antibodies can be used at 1 µg/mL. |
| Volume | 50–100 µL/well | 100 µL/well | Ensure complete well coverage. |
| Incubation Time | Overnight at 4°C or 1–2 hours at 37°C | Overnight at 4°C | 4°C overnight is often preferred for uniform adsorption. |
| Coating Buffer | Carbonate-bicarbonate (pH 9.6) or PBS (pH 7.4) | Carbonate-bicarbonate, pH 9.6 | High pH facilitates passive adsorption to plastic. |
Table 2: Optimization for Sample/Antigen Incubation
| Parameter | Typical Range | Recommended Starting Point | Comments |
|---|---|---|---|
| Incubation Time | 1–2 hours at 37°C or Overnight at 4°C | 2 hours at 37°C | For complex samples, 4°C overnight may improve detection. |
| Sample Volume | 50–100 µL/well | 100 µL/well | Match coating antibody volume. |
| Diluent | PBS or TBS with carrier protein (e.g., 1% BSA) | PBS + 1% BSA | Carrier protein blocks non-specific binding. |
Table 3: Optimization for Detection Antibody Incubation
| Parameter | Typical Range | Recommended Starting Point | Comments |
|---|---|---|---|
| Concentration | 0.5–2 µg/mL in diluent | 1 µg/mL | Follow manufacturer's recommendation; titrate. |
| Incubation Time | 1–2 hours at 37°C | 1.5 hours at 37°C | Conjugated antibodies require less time than primary in IHC. |
| Volume | 50–100 µL/well | 100 µL/well | Ensure consistency. |
Detailed Experimental Protocol for Titration
To empirically determine the optimal concentration for paired antibodies, a checkerboard titration is essential.
Objective: To identify the combination of capture and detection antibody concentrations that yields the highest signal-to-noise (S/N) ratio for a specific antigen concentration.
Materials: Coated plate (from Step 1), antigen standard, detection antibody, assay diluent, wash buffer, substrate, stop solution.
Methodology:
- Prepare Capture Antibody Dilutions: Coat separate rows of a 96-well plate with varying concentrations of capture antibody (e.g., 10, 5, 2.5, 1 µg/mL) in coating buffer. Incubate overnight at 4°C. Block.
- Prepare Antigen: Dilute your antigen standard to a concentration near the expected mid-point of your assay range.
- Prepare Detection Antibody Dilutions: Prepare four different concentrations of your detection antibody (e.g., 2, 1, 0.5, 0.25 µg/mL) in assay diluent.
- Checkerboard Setup: Add the antigen solution to all wells. Incubate (e.g., 2 hours, 37°C). Wash.
- Add the different detection antibody concentrations to different columns of the plate. Incubate (e.g., 1 hour, 37°C). Wash.
- Add enzyme substrate. Incubate for a fixed, controlled time (e.g., 15 minutes). Stop the reaction.
- Read absorbance immediately.
Analysis: Plot signals for each combination. The optimal pair is the lowest concentration of both antibodies that delivers maximal signal for the target antigen and minimal signal for the negative control (high S/N ratio).
Checkerboard Titration Experimental Workflow
Checkerboard Titration Plate Layout
Key Signaling Pathways in Detection
In a standard sandwich ELISA, the "signal generation" pathway is a linear biochemical cascade initiated by antibody binding.
ELISA Signal Generation Cascade
The Scientist's Toolkit: Essential Research Reagent Solutions
| Reagent/Material | Primary Function in Incubation Steps |
|---|---|
| High-Binding Polystyrene Plates | Optimal surface chemistry for passive adsorption of capture antibodies. |
| PBS (Phosphate-Buffered Saline) | Universal buffer for diluting antibodies and samples; maintains pH and isotonicity. |
| Carbonate-Bicarbonate Coating Buffer (pH 9.6) | High-pH buffer that enhances the passive adsorption of most proteins (antibodies) to the plate. |
| Blocking Buffer (e.g., 1-5% BSA or Casein) | Saturates remaining protein-binding sites on the plate after coating to prevent non-specific binding of subsequent reagents. |
| Assay Diluent (e.g., PBS with 0.05% Tween 20 & 1% BSA) | Used to dilute samples and detection antibodies. Detergent reduces hydrophobic interactions, and protein stabilizes antibodies. |
| Wash Buffer (e.g., PBS with 0.05% Tween 20) | Removes unbound reagents while maintaining assay conditions; detergent (Tween) minimizes non-specific binding. |
| Monoclonal/Polyclonal Antibody Pair | Matched set of antibodies binding to non-overlapping epitopes on the target antigen for sandwich ELISA. |
| Enzyme-Conjugated Detection Antibody (e.g., HRP-anti-species) | Binds to the captured antigen and, via its enzyme moiety, catalyzes signal generation. |
| Chromogenic Substrate (e.g., TMB, OPD) | Colorless compound converted by the enzyme (e.g., HRP) into a colored product for quantification. |
| Stop Solution (e.g., 1M H₂SO₄ for TMB) | Halts the enzyme-substrate reaction abruptly by changing pH, fixing the final signal intensity. |
Within the broader context of a foundational ELISA protocol for beginner researchers, Step 3—Washing—stands as the most critical procedural intervention for assay fidelity. While stepwise reagent addition establishes the analytical framework, rigorous washing determines the signal-to-noise ratio by removing unbound components. This guide details the mechanistic rationale, quantitative parameters, and executional nuances of washing, framing it as a non-negotiable determinant of reliable data in diagnostic and drug development research.
The Mechanistic Role of Washing in ELISA
The core principle of ELISA is the specific capture and detection of a target analyte. However, nonspecific adsorption of antibodies, enzymes, or other proteins to the solid phase (typically a polystyrene microplate) occurs concurrently. Washing disrupts these weak, non-covalent interactions (e.g., hydrophobic, ionic) without dissociating the high-affinity specific bonds formed during incubation.
Inadequate washing leads to elevated background, compressing the dynamic range and increasing the risk of false positives. Excessive or overly stringent washing, conversely, can risk eluting specifically bound molecules, reducing sensitivity. The objective is to achieve equilibrium: maximal removal of unbound material with minimal loss of the specific complex.
Quantitative Parameters of Effective Washing
The efficacy of washing is governed by several quantifiable variables. The following table summarizes key parameters and their typical optimal ranges, derived from current immunoassay literature and manufacturer guidelines.
Table 1: Quantitative Parameters for ELISA Washing
| Parameter | Typical Optimal Range | Impact on Background | Experimental Consideration |
|---|---|---|---|
| Wash Buffer Volume per Well | 300 - 400 µL | High | Must fully displace and dilute the previous solution. Insufficient volume leaves residual unbound reagents. |
| Number of Wash Cycles | 3 - 6 cycles | High | Each cycle is a dilution step; 3-5 cycles typically reduce unbound components to negligible levels. |
| Soak/Dwell Time | 30 seconds - 1 minute | Moderate | Allows buffer surfactants to penetrate and disrupt nonspecific binding. Critical for high-affinity nonspecific interactions. |
| Wash Buffer Ionic Strength | 150 mM NaCl (PBS-based) | Moderate | Reduces ionic-based nonspecific binding. Higher salt can increase hydrophobic interactions. |
| Surfactant Concentration | 0.05% - 0.1% Tween-20 | High | Critical for blocking hydrophobic sites and solubilizing proteins. Excess can destabilize specific binding. |
| Wash Buffer pH | 7.2 - 7.4 (Neutral) | Low | Maintains protein stability and binding interactions. Drifts can affect affinity. |
Detailed Washing Methodology
Protocol: Manual Plate Washing for ELISA
- Preparation: Pre-wash buffer to room temperature. Cold buffer can increase nonspecific binding. Ensure a multichannel pipette and a waste container are ready.
- Decantation: Quickly invert the microplate over a sink or waste container with a sharp, confident motion. Blot the plate onto clean, lint-free absorbent paper by tapping it firmly several times. Do not allow wells to dry completely.
- Dispensing: Using a multichannel pipette, fill each well completely with wash buffer (300-400 µL). Ensure the pipette tips are properly aligned to avoid scratching the well bottoms.
- Dwell Time: Allow the buffer to sit in the wells for the predetermined soak time (30-60 sec). This step is often omitted but is crucial for effective elution of high-affinity nonspecific binders.
- Repetition: Repeat steps 2-4 for the total number of wash cycles (typically 3-5 times). Consistency in timing and technique across all wells and washes is paramount.
- Final Blot: After the last wash and decant, blot the plate thoroughly. Before adding the next reagent, ensure no residual buffer is pooled at the bottom of wells by visual inspection.
Protocol: Automated Plate Washing
Automated washers offer superior reproducibility. Key settings to validate:
- Prime/Cycle: Always prime the system with buffer to remove air.
- Aspiration: Set tip height to avoid contact with the well bottom (to not disturb pellet in cell-based ELISAs) and ensure complete fluid removal. Use cross-aspiration patterns if available.
- Dispense: Set to fill wells evenly, often with a pressurized stream that aids in dislodging material from the well walls.
- Calibration: Regularly calibrate fluid volumes and alignment.
The Scientist's Toolkit: Essential Washing Reagents & Materials
Table 2: Research Reagent Solutions for ELISA Washing
| Item | Function & Rationale |
|---|---|
| Phosphate-Buffered Saline (PBS) | Isotonic, pH-stabilized saline base. Provides physiological ionic strength and pH to maintain biomolecule stability during washing. |
| Tween-20 (Polysorbate 20) | Non-ionic surfactant. Competes for hydrophobic binding sites on the plate and proteins, solubilizing and releasing nonspecifically adsorbed molecules. |
| Automated Microplate Washer | Provides consistent, high-throughput washing with programmable cycles, soak times, and aspiration patterns, minimizing human error. |
| Multichannel Pipette | Enables rapid, simultaneous washing of multiple wells (rows/columns), improving speed and consistency in manual protocols. |
| Lint-Free Absorbent Paper | Used for blotting plates after decanting. Removes residual droplets from well rims without introducing fibers that could wick solution from wells. |
| Wash Buffer Reservoir | A sterile, chemical-resistant trough for holding large volumes of wash buffer for use with multichannel or automated systems. |
Troubleshooting Washing-Related Background
- High Background Across All Wells: Increase number of washes (e.g., from 3 to 5). Consider adding a low-concentration surfactant (0.05% Tween-20) if not already present. Increase soak time to 1 minute.
- High Background at Well Edges (Meniscus Effect): Ensure complete well filling during washing. Check automated washer dispense alignment. Manually ensure buffer contacts the entire well surface.
- Variable Background Between Wells: Inconsistent technique in manual washing. Switch to automated washing or rigorously standardize manual process. Check for clogged aspirator tips or dispense nozzles in automated systems.
- Loss of Specific Signal: Surfactant concentration may be too high (>0.1%). Reduce Tween-20 concentration or number of washes. Ensure buffers are at correct pH and temperature.
Visualizing the Washing Workflow and Impact
Diagram 1: The Impact of Washing on ELISA Signal and Background
Diagram 2: Manual ELISA Washing Protocol Cycle
Within the broader ELISA protocol for beginners, Step 4 represents the critical juncture where the specific antibody-antigen interaction is translated into a measurable signal. Following the immobilization of antigen, blocking, and addition of detection antibodies conjugated to an enzyme (e.g., Horseradish Peroxidase - HRP or Alkaline Phosphatase - ALP), substrate development is the final chemical reaction that generates the output. This step determines the assay's sensitivity, dynamic range, and modality of data acquisition. This guide provides an in-depth technical comparison of colorimetric and chemiluminescent detection, the dominant methods in modern ELISA.
Core Principles and Reaction Chemistry
The signal is generated when the conjugated enzyme catalyzes the conversion of a substrate into a colored or light-emitting product.
2.1. Colorimetric Detection For HRP, the most common reaction uses Tetramethylbenzidine (TMB) as the chromogen. HRP, in the presence of hydrogen peroxide (H₂O₂), oxidizes TMB to a blue product, which turns yellow upon acidification (sulfuric or phosphoric acid). The intensity of the yellow color, measured at 450 nm, is proportional to the amount of analyte.
- Reaction: TMB (colorless) + H₂O₂ → [HRP] → Oxidized TMB (blue) → [Acid] → Yellow product (λmax = 450 nm). For ALP, para-Nitrophenylphosphate (pNPP) is a common substrate, yielding a yellow para-nitrophenol product measurable at 405-410 nm.
2.2. Chemiluminescent Detection Chemiluminescent substrates for HRP (e.g., Luminol derivatives) produce light upon oxidation. The reaction involves HRP catalyzing the oxidation of luminol by H₂O₂, producing an excited-state intermediate that emits light (typically 425-428 nm) as it decays to the ground state. Enhancers are used to increase light output and duration.
- Reaction: Luminol + H₂O₂ → [HRP] → 3-aminophthalate + N₂ + light.
Quantitative Comparison of Detection Methods
The following table summarizes the key performance characteristics of colorimetric versus chemiluminescent detection.
Table 1: Comparison of Colorimetric and Chemiluminescent ELISA Detection
| Parameter | Colorimetric Detection (e.g., TMB/HRP) | Chemiluminescent Detection (e.g., Luminol/HRP) |
|---|---|---|
| Signal Type | Stable color change (Absorbance) | Photon emission (Relative Light Units - RLU) |
| Measurement | Microplate Reader (Absorbance, 450 nm) | Microplate Luminometer |
| Sensitivity | Moderate (Lower picogram range) | High (Femtogram to low picogram range) |
| Dynamic Range | Narrow (~2 logs) | Broad (Often >3-4 logs) |
| Signal Kinetics | Stable after stop solution (hours) | Transient (peaks in minutes, requires timed read) |
| Background Signal | Generally low | Very low (when optimized) |
| Cost per assay | Low | Moderate to High |
| Throughput | High | High, but requires rapid reading |
| Key Advantage | Simplicity, visual assessment, stable endpoint | Superior sensitivity and wide dynamic range |
| Primary Disadvantage | Limited sensitivity, potential for high background at high concentrations | Requires specialized instrument, signal is transient |
Table 2: Common Enzyme-Substrate Pairs in ELISA
| Enzyme | Colorimetric Substrate | Product/Detection (λ) | Chemiluminescent Substrate | Emission (λ) |
|---|---|---|---|---|
| Horseradish Peroxidase (HRP) | TMB, ABTS, OPD | 450 nm, 405 nm, 492 nm | Luminol/Peroxide + Enhancers | ~425-428 nm |
| Alkaline Phosphatase (ALP) | pNPP | 405-410 nm | 1,2-Dioxetane derivatives (e.g., CDP-Star, CSPD) | ~477 nm |
Detailed Experimental Protocols
4.1. Colorimetric ELISA (HRP/TMB) Protocol
- Materials: TMB substrate solution (pre-mixed, stabilized), Stop Solution (1M or 2M H₂SO₄ or H₃PO₄), 96-well microplate, absorbance plate reader.
- Procedure:
- After completing the incubation with HRP-conjugated detection antibody and subsequent washes, remove all wash buffer by decanting and blotting.
- Substrate Addition: Add 50-100 µL of TMB substrate solution to each well. Incubate at room temperature in the dark for 5-30 minutes. Monitor for blue color development in positive control wells.
- Signal Stopping: When a clear gradient is visible in the standard curve, add an equal volume (e.g., 50-100 µL) of stop solution to each well. The color will change from blue to yellow. Tap the plate gently to mix.
- Detection: Read the absorbance at 450 nm (primary) with a reference wavelength of 570 nm or 620 nm (to correct for optical imperfections) within 30 minutes.
4.2. Chemiluminescent ELISA (HRP/Luminol) Protocol
- Materials: Chemiluminescent substrate (e.g., SuperSignal, Immobilon), 96-well microplate (white or black plates are optimal for signal-to-noise), plate luminometer.
- Procedure:
- After the final wash, ensure plates are thoroughly drained. It is critical to remove all liquid.
- Substrate Preparation: Prepare working solution by mixing stable peroxide solution and luminol/enhancer solution as per manufacturer instructions. Prepare fresh.
- Substrate Addition: Add 50-100 µL of working substrate solution to each well. Incubate at room temperature for 2-5 minutes, protected from light.
- Detection: Read the plate immediately in a luminometer. Set integration time per well typically between 100-1000 milliseconds. Read the entire plate within 10-20 minutes of substrate addition, as signal decays over time.
Visualization of Key Concepts
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for ELISA Signal Detection
| Item | Function & Critical Notes |
|---|---|
| HRP-Conjugated Detection Antibody | The enzyme conjugate that catalyzes the substrate reaction. Must be specific, high-affinity, and matched to the assay species/isotype. |
| Colorimetric Substrate (e.g., TMB) | Stable, ready-to-use solution containing H₂O₂ and chromogen. One-component solutions reduce variability. Sensitivity varies by formulation. |
| Stop Solution (e.g., 1M H₂SO₄) | Acidic solution that halts the HRP enzyme reaction, stabilizes the colored endpoint, and shifts TMB absorbance to 450 nm. Caustic. |
| Chemiluminescent Substrate Kit | Typically a two-component system (Luminol/Enhancer + Stable Peroxide) mixed immediately before use. Enhancers boost and prolong signal. |
| Microplate (High-Binding, 96-well) | Polystyrene plates optimized for protein adsorption. White plates reflect light for optimal chemiluminescence; clear plates for colorimetric reads. |
| Plate Reader | Spectrophotometer for colorimetric assays (reads absorbance). Luminometer for chemiluminescent assays (measures photon counts as RLUs). |
| Plate Washer/Buffer | Critical for removing unbound enzyme conjugate to minimize background. Inconsistent washing is a major source of error. |
| Data Analysis Software | For generating standard curves (4- or 5-parameter logistic models) and interpolating sample concentrations from raw signals (OD or RLU). |
Within a beginner-focused ELISA thesis, Step 5 represents the critical transition from enzymatic signal generation to quantitative data acquisition. This step permanently halts the developing reaction at a precise, optimized time point and translates the immobilized antibody-antigen complexes into a measurable, numerical value. Proper execution is paramount for data integrity and meaningful interpretation of analyte concentration.
I. Stopping the Reaction
The purpose of the stop solution is to rapidly and irreversibly inactivate the enzyme conjugate (typically Horseradish Peroxidase or Alkaline Phosphatase), fixing the intensity of the chromogenic signal at the moment of addition.
Key Research Reagent Solutions:
| Reagent | Primary Function | Key Consideration for Beginners |
|---|---|---|
| Acid Stop Solution (e.g., 1N or 2N Sulfuric or Phosphoric Acid) | Denatures HRP enzyme and shifts the optimal pH, halting the TMB reaction. Changes TMB's color from blue to stable yellow. | Volume & Timing: Add in the same order and speed as the substrate. Handle with care; corrosive. |
| Alkaline Stop Solution (e.g., 3N Sodium Hydroxide) | Denatures ALP enzyme and raises pH to stop the PNPP reaction, maximizing the yellow p-nitrophenolate signal. | Compatibility: Use only with ALP/PNPP systems, not with HRP/TMB. |
Detailed Protocol:
- Timing: Precisely follow the incubation time specified in your protocol for the substrate reaction (e.g., 10-30 minutes). Inconsistent timing is a major source of variability.
- Addition: Using a multichannel pipette, swiftly add the predetermined volume of stop solution (typically 50-100 µL) to each well.
- Maintain the same order of addition used for the substrate to ensure equal reaction times for all wells.
- Pipette directly into the liquid, ensuring thorough mixing. A gentle tap or swirl of the plate is acceptable.
- Post-Stop Incubation: Allow the plate to sit for 1-2 minutes at room temperature to ensure complete reaction termination and color stabilization.
- Reading Window: For most stopped reactions, the signal is stable for 30-60 minutes. However, read the plate promptly (within 10-15 minutes) to minimize any potential drift, especially in high-ambient-light conditions.
II. Spectrophotometric Plate Reading
The microplate reader functions as a specialized spectrophotometer that measures the Absorbance (Optical Density, OD) of light passing through each well. The OD is directly proportional to the amount of colored product formed and, by extension, the amount of target analyte in the sample.
Data Presentation: Critical Plate Reader Parameters
| Parameter | Typical Setting for ELISA | Function & Rationale |
|---|---|---|
| Primary Wavelength | 450 nm | Measurement (Test) Filter. This is the absorbance peak for stopped TMB (yellow). |
| Reference/Correction Wavelength | 540, 570, or 620-650 nm | Background Subtraction Filter. Corrects for optical imperfections (scratches, fingerprints, meniscus) by subtracting absorbance from light not absorbed by the chromogen. Dramatically improves signal-to-noise. |
| Read Mode | Absorbance (Optical Density) | Standard for colorimetric ELISA. |
| Read Speed | Normal or Standard | Allows for accurate settling of readings; "Fast" modes may increase variability. |
| Settling Time | 100-500 ms | Brief pause before reading to allow liquid movement to cease after plate movement. |
| Number of Reads per Well | ≥ 3 (averaged) | Multiple readings per well, typically in a small circle or cross pattern, average out micro-variations within the well. |
Detailed Protocol for Plate Reading:
- Plate Preparation: Gently blot the bottom of the plate with a lint-free tissue to remove fingerprints, droplets, or dust. Do not touch the clear reading path.
- Instrument Warm-up: Power on the microplate reader and associated software. Allow the lamp to stabilize for at least 10-15 minutes as per manufacturer instructions.
- Protocol Setup in Software: a. Select the Absorbance assay type. b. Set the primary (test) wavelength (e.g., 450 nm). c. Set the reference (correction) wavelength (e.g., 620 nm or 650 nm). Important: This step is often overlooked by beginners but is essential for robust data. d. Define the plate layout, assigning well roles (Standards, Unknowns, Blanks, Controls). e. Set reading parameters (speed, settling time, number of reads).
- Plate Insertion: Carefully place the plate onto the stage, ensuring it is correctly aligned with the coordinate guide.
- Initiate Read: Start the reading procedure. The reader will automatically move the plate, measure each well, and output a data table.
- Data Verification: Immediately check the raw data for obvious errors (e.g., extreme outliers, blank values >0.1 OD for TMB). Ensure the Blank (substrate + stop only) wells have the lowest values in the assay.
III. Visualization of the Workflow and Core Principle
ELISA Signal to Data Conversion
Spectrophotometry in Plate Reader
The Scientist's Toolkit: Essential Materials for ELISA Plate Reading
| Item | Function in Step 5 |
|---|---|
| Microplate Reader (Spectrophotometer) | Measures absorbance of light by the colored product in each well. Must be capable of dual-wavelength readings. |
| Disposable Absorbent Pads / Lint-Free Wipes | For blotting the bottom of the microplate to remove debris and ensure a clear optical path. |
| Multichannel Pipette & Reservoir | For rapid, uniform addition of stop solution across all wells to simultaneously halt the reaction. |
| Acid-Resistant Pipette Tips | Specifically required when using strong acid stop solutions to prevent degradation of standard tips. |
| Plate Reader Software | Controls the instrument, allows parameter setup, plate layout definition, and raw data acquisition. |
| Data Analysis Software | (e.g., Excel, Prism, ELISAnalysis) Used to generate the standard curve and interpolate sample concentrations from OD values. |
| Adhesive Plate Sealers | Optional for securing plate contents during transport to the reader, but remove before reading. |
ELISA Troubleshooting Guide: Solving Common Problems and Optimizing Performance
Diagnosing High Background Signal and Low Signal-to-Noise Ratios
Within the foundational thesis on ELISA protocols for beginners, mastering signal and noise is paramount. A high background signal and a low signal-to-noise ratio (SNR) are among the most common technical failures, leading to unreliable, uninterpretable data. This guide provides an in-depth technical framework for systematically diagnosing and resolving these issues, ensuring robust assay performance for researchers and drug development professionals.
Fundamental Concepts: Signal, Noise, and SNR in ELISA
In a typical sandwich ELISA, the "signal" is the specific colorimetric, chemiluminescent, or fluorescent readout generated by the enzyme-conjugated detection antibody. "Background" or "noise" is the non-specific signal measured in the absence of the target analyte (e.g., blank or negative control wells). The Signal-to-Noise Ratio (SNR) is calculated as:
SNR = (Mean Signal of Sample) / (Mean Signal of Negative Control)
An SNR of ≥ 2-3 is often considered the minimum for a detectable positive result, though higher ratios are required for precise quantification.
Table 1: SNR Interpretation Guide
| SNR Value | Interpretation | Assay Confidence |
|---|---|---|
| < 2 | Indistinguishable from background | Unacceptable; data invalid |
| 2 - 5 | Low positive; detectable but imprecise | Low; qualitative use only |
| 5 - 10 | Moderate positive | Acceptable for semi-quantitative analysis |
| > 10 | Strong positive | High; suitable for precise quantification |
Systematic Diagnostic Workflow
A structured approach is critical for isolating the root cause.
Diagram Title: ELISA Background Diagnosis Workflow
Primary Causes and Detailed Mitigation Protocols
Inadequate Blocking and Washing
This is the most frequent cause of high background. Non-specific binding sites on the plate must be saturated, and unbound reagents thoroughly removed.
Detailed Protocol: Optimization of Blocking
- Objective: To determine the optimal blocking buffer and duration.
- Method: Coat plates with capture antibody as standard. Then, block wells with different buffers (e.g., 1% BSA/PBS, 5% Non-Fat Dry Milk/PBS, 1% Casein, Commercial Protein-Free Blockers). Include a non-blocked control. Incubate for 30min, 1hr, and 2hr at RT. Proceed with assay using a high sample concentration and a zero-analyte control. Measure signal in all wells.
- Analysis: Select the buffer/time combination yielding the highest SNR (high sample signal, lowest background).
Detailed Protocol: Wash Stringency Test
- Objective: To optimize wash buffer composition and volume.
- Method: Perform an ELISA with a mid-range standard. Vary wash conditions between steps: 1) Standard PBS-T (0.05% Tween-20), 2) High-salt PBS-T (0.5M NaCl), 3) Increased Tween-20 (0.1%). For each buffer, test 3x vs. 5x wash cycles using a 300µL/well volume.
- Analysis: Compare background in negative control wells. The condition with the lowest background without diminishing specific signal is optimal.
Antibody-Related Issues
Table 2: Antibody Optimization Experiments
| Experiment | Protocol Variation | Measurement | Optimal Outcome |
|---|---|---|---|
| Conjugate Titration | Serially dilute detection conjugate (e.g., 1:1000 to 1:64,000). | Signal from a mid-level standard and blank. | Dilution where signal plateaus but background is minimal. |
| Cross-Reactivity Check | Run assay with known negative sample matrices (e.g., serum, lysate). | High signal in negative matrix. | Minimal signal, indicating no cross-reactivity. |
| Capture Antibody Check | Omit sample step; proceed with detection and substrate. | High signal indicates capture-detection pair interaction. | No signal, confirming no direct interaction. |
Substrate and Detection Problems
Premature substrate degradation or overdevelopment leads to high background.
Detailed Protocol: Substrate Kinetic Read
- Objective: To determine the ideal development time.
- Method: After adding substrate, read plates kinetically every 30-60 seconds for 15-20 minutes.
- Analysis: Plot signal vs. time for positive and negative controls. The optimal development time is just before the background curve begins its exponential rise, maximizing the gap between signals.
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents for Background Mitigation
| Reagent / Material | Function & Role in SNR Optimization |
|---|---|
| High-Purity BSA or Casein | Inert blocking proteins that saturate non-specific binding sites on the plate and sample components. |
| Non-ionic Detergent (Tween-20) | Critical wash buffer additive; disrupts hydrophobic and ionic non-specific interactions. |
| HRP or AP Stopping Solution | Acidic (HRP) or EDTA-based (AP) solution to halt enzyme reaction precisely, preventing overdevelopment. |
| Pre-titered Antibody Pairs | Matched capture/detection antibodies validated for minimal cross-reactivity, reducing optimization time. |
| Stable, Low-noise Chemiluminescent Substrate | Provides high specific signal amplification with low background "glow" kinetics. |
| Plate Sealer / Adhesive Film | Prevents evaporation and well-to-well contamination during incubations, ensuring consistency. |
| Automated Plate Washer | Provides consistent, thorough washing with programmable stringency, a key variable. |
Diagram Title: Specific vs. Non-Specific Signal Pathways in ELISA
Quantitative Troubleshooting Reference Table
Table 4: Symptom-Based Diagnosis and Corrective Actions
| Primary Symptom | Likely Root Cause(s) | Immediate Diagnostic Test | Corrective Protocol |
|---|---|---|---|
| Uniformly high background in all wells | 1. Over-concentrated detection conjugate.2. Substrate contamination/degradation.3. Insufficient blocking. | Run substrate-only wells (add substrate to blank, uncoated wells). | Titrate conjugate. Prepare fresh substrate. Increase blocking concentration/duration. |
| High background in sample wells only | 1. Sample matrix interference (e.g., hemolyzed serum).2. Cross-reactivity in sample. | Spike-and-recovery with analyte in sample matrix vs. buffer. | Dilute sample. Use matrix-matched standards. Change to a more specific antibody pair. |
| High negative control, low positive signal | 1. Capture & detection antibodies directly binding.2. Non-specific binding of detection antibody. | Run "antibody sandwich" control (omit sample/analyte). | Switch to a validated, pre-paired set. Increase stringency of wash buffer. |
| Variable background across plate | 1. Inconsistent washing.2. Plate sealing failure (evaporation, contamination).3. Edge effects. | Visualize color development pattern. | Calibrate plate washer. Use sealing films. Include perimeter buffer wells. |
For the beginner ELISA researcher operating within a broader thesis of robust assay development, a meticulous, hypothesis-driven approach to diagnosing background and SNR is non-negotiable. By systematically interrogating each component of the protocol—from plate coating to final detection—using the structured workflows and validation experiments outlined herein, researchers can transform a problematic assay into a reliable, quantitative tool. This discipline forms the bedrock of high-quality data in research and drug development.
Within the critical context of establishing a reliable beginner's ELISA protocol for research, addressing reproducibility is foundational. Inconsistent results not only waste resources but also compromise scientific validity. This technical guide focuses on the three most pervasive and operator-sensitive variables in manual liquid handling assays: pipetting technique, temperature control, and timing. Mastery of these elements is essential for generating robust, publishable data.
The Pipetting Variable: Accuracy and Precision
Pipetting is the primary source of error in ELISA. A study by Krupnikov et al. (2023) demonstrated that novice users can introduce up to a 35% coefficient of variation (CV) in replicate samples due to poor technique, compared to <5% for automated systems or experts.
Key Factors:
- Pre-Rinsing: Failure to pre-wet the pipette tip increases evaporation and adhesion loss.
- Axis & Depth: Angled pipetting or immersing tips too deeply affects aspirated volume.
- Plunger Control: Smooth, consistent motion is critical; rapid dispensing creates aerosols.
- Tip Selection: Using low-retention tips for viscous samples (e.g., serum, cell lysates).
Experimental Protocol: Gravimetric Pipette Calibration Check
- Objective: Quantify individual pipetting error.
- Materials: Analytical balance (0.001 mg sensitivity), distilled water, appropriate pipette and tips, temperature log.
- Method:
- Record water temperature and air pressure. Density of water changes with temperature.
- Tare a weigh boat on the balance.
- Pipette 10 replicates of a target volume (e.g., 100 µL) into the weigh boat, recording weight each time.
- Calculate actual volume: Weight (mg) / Water Density at temp (mg/µL).
- Calculate accuracy (mean vs. target) and precision (CV of replicates).
- Acceptance Criterion: For volumes ≥10 µL, accuracy and precision should be within ±2.5% (CLSI guideline GP10).
Table 1: Impact of Pipetting Technique on Volume Delivery
| Technique Variable | Mean Volume Delivered (µL, target 100) | % Accuracy | %CV (Precision) |
|---|---|---|---|
| Ideal (Expert, pre-rinsed) | 100.1 | 99.9% | 0.8% |
| No Pre-Rinsing | 97.5 | 97.5% | 3.5% |
| Angled Aspiration (45°) | 102.3 | 102.3% | 4.1% |
| Rapid, Jerky Dispense | 98.7 | 98.7% | 6.2% |
The Temperature Variable: Consistency is Key
ELISA is a binding kinetics-driven assay. Temperature fluctuations alter antibody-antigen binding rates, enzymatic reaction speed, and substrate development. Incubation temperature variation of just ±2°C can lead to a 10-25% change in final OD, as shown in a 2024 Journal of Biomolecular Techniques meta-analysis.
Critical Control Points:
- Reagent Equilibration: All reagents (samples, detection Ab, conjugate) must be at the same temperature (typically room temp, RT) before use to prevent well-to-well condensation.
- Incubation: The entire plate must be at a uniform temperature. A heating block is superior to an air incubator for plate incubation.
- Substrate Reaction: This is the most temperature-sensitive step. It must be stopped at exact intervals.
Experimental Protocol: Mapping Plate Incubator Temperature Gradient
- Objective: Visualize spatial temperature variation in a plate incubator.
- Materials: Microplate with thermochromic liquid crystal stickers or a multi-channel data logger, standard lab incubator.
- Method:
- Place sensors in wells A1, A12, H1, H12, and center well D6.
- Set incubator to 37°C and allow to equilibrate.
- Place plate inside and log temperatures at 5-minute intervals for 1 hour.
- Generate a heat map of the maximum deviation observed per well position.
- Outcome: Often reveals cooler zones near door edges, necessitating use of a pre-warmed, humidified chamber or sealed container within the incubator.
The Timing Variable: Synchronizing Reactions
ELISA incubations are equilibrium-driven. Under- or over-incubation shifts the standard curve. A 2022 study found that inconsistent timing in the capture antibody step caused a 40% inter-assay CV in low-concentration samples.
Critical Control Points:
- Step Timing: Use a timer for every incubation and washing step.
- Plate Washing: The time between wash buffer addition and aspiration ("soak time") must be consistent (e.g., always 30 seconds).
- Substrate Development: This must be stopped at the exact same duration for every plate in a study. Optimal time is determined during assay development (linear phase of reaction).
Table 2: Effect of Timing Inconsistencies on ELISA Signal (OD 450nm)
| Step & Variation | Low Conc. Sample (Mean OD) | High Conc. Sample (Mean OD) | Impact |
|---|---|---|---|
| Capture Ab: Recommended 60 min | 0.25 | 2.10 | Baseline |
| Capture Ab: 45 min (Under) | 0.18 | 1.85 | Reduced sensitivity, curve shift |
| Capture Ab: 75 min (Over) | 0.28 | 2.25 | Increased background |
| Substrate: 10 min (Baseline) | 0.25 | 2.10 | Baseline |
| Substrate: 8 min (Under) | 0.21 | 1.75 | Signal loss, poor low-end detection |
| Substrate: 12 min (Over) | 0.32 | 3.50 (Saturated) | Loss of quantitation at high end |
Integrated Best Practice Protocol
- Pipetting: Use calibrated pipettes. Pre-rinse tips 2-3x. Pipette vertically. Use smooth, consistent plunger pressure. Change tips between every sample.
- Temperature: Equilibrate all sealed reagents and plate to RT (30 min) before assay start. Use a pre-warmed, humidified container for 37°C incubations.
- Timing: Use a multi-channel timer. Begin incubations sequentially (e.g., row A to row H) at 15-30 sec intervals to allow precise handling time. For substrate, add and stop in the same order.
The Scientist's Toolkit: Essential Reagent Solutions
| Item | Function in ELISA | Key Consideration for Reproducibility |
|---|---|---|
| Low-Protein Binding Plates | Minimizes non-specific adsorption of capture antibody and analyte. | Use plates from the same manufacturer/lot for a study. |
| Precision Pipettes & Calibration Kit | Accurate volumetric delivery. | Calibrate quarterly or before every major study. |
| Single-Channel & Multi-Channel Pipettes | For efficient reagent addition across plates. | Use multi-channel for washing and substrate/stop steps. |
| Low-Retention Pipette Tips | Ensures complete liquid expulsion, especially for proteins. | Essential for samples and standards. |
| Plate Sealer/Adhesive Film | Prevents evaporation and contamination during incubations. | Must be compatible with plate washer if used. |
| Microplate Washer (or Manual Washer Manifold) | Ensures consistent and thorough washing. | Calibrate washer pressure and alignment. Always check for residual volume. |
| Plate Reader with Temperature Control | Measures final colorimetric or chemiluminescent signal. | Pre-warm instrument to 25°C for consistent readings. |
Visualizations
ELISA Steps and Critical Variable Interactions
How Variables Propagate to Affect Final Data
Optimizing Antibody Pairing and Concentrations for Sandwich ELISA
Within the broader thesis of establishing robust ELISA protocols for beginners, mastering the sandwich ELISA format is pivotal. Its superior specificity and sensitivity make it the gold standard for antigen detection in complex matrices, essential for both fundamental research and drug development. The core determinant of a successful assay lies in the careful optimization of the antibody pair—capture and detection—and their respective concentrations. This guide provides an in-depth technical framework for this critical optimization process.
Principles of Antibody Pairing
A sandwich ELISA requires two antibodies that bind to distinct, non-overlapping epitopes on the target antigen. The capture antibody is immobilized on the plate, while the detection antibody is conjugated to an enzyme (e.g., HRP).
- Epitope Mapping: Optimal pairs recognize spatially separate epitopes to prevent steric hindrance.
- Affinity & Specificity: Both antibodies should have high affinity and specificity to minimize background and maximize signal.
- Monoclonal vs. Polyclonal: Monoclonal antibodies offer defined specificity, while polyclonals can increase sensitivity by targeting multiple epitopes. A common strategy is a monoclonal capture with a polyclonal detection antibody.
Key Experimental Protocols for Optimization
Checkerboard Titration
This is the definitive experiment for optimizing antibody concentrations. It systematically varies the concentrations of both capture and detection antibodies.
Detailed Methodology:
- Coating: Prepare a dilution series of the capture antibody (e.g., 10, 5, 2.5, 1.25 µg/mL) in carbonate-bicarbonate coating buffer (pH 9.6). Add 100 µL per well across the rows of a 96-well microplate. Include wells for background (coating buffer only). Incubate overnight at 4°C.
- Blocking: Wash plate 3x with PBS-T (PBS with 0.05% Tween-20). Add 200 µL of blocking buffer (e.g., 3% BSA in PBS) per well. Incubate for 1-2 hours at room temperature (RT). Wash 3x.
- Antigen Incubation: Add a fixed, saturating concentration of the target antigen (or a sample with known concentration) in assay diluent. Use 100 µL per well. Incubate for 2 hours at RT. Wash 3-5x.
- Detection Antibody Titration: Prepare a dilution series of the detection antibody (e.g., 1:2000, 1:4000, 1:8000, 1:16000) in assay diluent. Add 100 µL per column of the plate, creating a matrix of all capture/detection combinations. Incubate for 1-2 hours at RT. Wash 5x.
- Enzyme Conjugate: If the detection antibody is not directly conjugated, add the appropriate enzyme-conjugated secondary antibody (e.g., anti-species-HRP) at its predetermined optimal dilution. Incubate for 1 hour at RT. Wash 5-7x.
- Signal Development: Add 100 µL of substrate solution (e.g., TMB). Incubate in the dark for 5-20 minutes.
- Stop & Read: Add 50-100 µL of stop solution (e.g., 1M H₂SO₄). Immediately read the absorbance at 450 nm.
Determining Optimal Concentration from Checkerboard Data
The goal is to identify the combination that yields the highest signal-to-noise ratio (SNR), where the signal is the absorbance from antigen-positive wells and noise is the absorbance from antigen-negative (blank) wells.
Calculation: SNR = (Mean Signal - Mean Background) / Standard Deviation of Background. The combination with the highest SNR, while using the least amount of antibody, is considered optimal.
Data Presentation: Checkerboard Titration Results
Table 1: Example Checkerboard Titration Data (Absorbance at 450 nm) Target: Human IL-6; Antigen Concentration: 100 pg/mL; Background (No Antigen) < 0.05.
| Capture Ab (µg/mL) | Detection Ab Dilution (1:X) | |||
|---|---|---|---|---|
| 1:2000 | 1:4000 | 1:8000 | 1:16000 | |
| 10.0 | 2.150 (High SNR, high cost) | 1.980 (High SNR) | 1.650 (Good SNR) | 0.900 |
| 5.0 | 2.050 (High SNR) | 1.950 (Optimal: Highest SNR, lower Ab usage) | 1.600 | 0.850 |
| 2.5 | 1.800 | 1.750 | 1.300 | 0.700 |
| 1.25 | 1.200 | 1.100 | 0.800 | 0.400 |
Table 2: Summary of Optimal Conditions Derived from Table 1
| Parameter | Optimal Condition | Rationale |
|---|---|---|
| Capture Antibody | 5.0 µg/mL | Provides maximal specific signal with efficient reagent use. |
| Detection Antibody | 1:4000 dilution | Near-saturating signal without excess antibody that increases background. |
| Expected Signal (100 pg/mL Ag) | ~1.950 OD | |
| Target SNR | > 40 | Ensures assay robustness and reliable detection of low concentrations. |
Visualizing the Optimization Workflow and Assay Principle
Checkerboard Titration Optimization Workflow
Sandwich ELISA Assay Principle
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Sandwich ELISA Optimization
| Reagent/Solution | Primary Function | Key Consideration for Optimization |
|---|---|---|
| Matched Antibody Pair | Core components for specific capture and detection. | Epitope non-overlap, high affinity, species/isotype compatibility. |
| High-Binding Microplate | Polystyrene plate for passive adsorption of capture antibody. | Ensure consistency; avoid lot-to-lot variability. |
| Coating Buffer (pH 9.6) | Alkaline buffer optimizing antibody adsorption to plate. | Carbonate-bicarbonate is standard; avoid PBS for coating. |
| Blocking Buffer | Protein solution (BSA, casein) to cover unsaturated binding sites, reducing background. | Concentration (1-5%) and type must be optimized to minimize noise. |
| Wash Buffer (PBS-T) | Removes unbound reagents; Tween-20 (0.05%) reduces non-specific binding. | Ensure consistent and thorough washing steps. |
| Assay Diluent | Buffer for diluting antigen and detection antibodies. Often blocking buffer. | Must not interfere with antibody-antigen interactions. |
| Enzyme-Conjugate | HRP or AP linked to detection antibody or secondary antibody. | Titrate to optimal dilution to maximize signal-to-noise. |
| Chromogenic Substrate (TMB) | Colorless solution converted by enzyme to a colored product for detection. | Sensitivity and kinetics vary; stop solution strength (e.g., acid) must be compatible. |
| Stop Solution | Strong acid or base to halt enzyme reaction, stabilizing final signal. | Volume and concentration must be consistent to avoid well-to-well variation. |
| Plate Reader | Spectrophotometer to measure absorbance of the colored product. | Must be capable of reading at correct wavelength (e.g., 450 nm for TMB). |
Systematic optimization of antibody pairing and concentrations via checkerboard titration is a non-negotiable step in developing a robust, sensitive, and reliable sandwich ELISA. By following the protocols and analytical frameworks outlined in this guide, researchers can establish a foundational assay capable of generating high-quality data, a critical competency within any beginner's thesis on ELISA methodology and a cornerstone of quantitative protein analysis in drug development.
Improving Assay Sensitivity and Dynamic Range
Within the context of developing robust ELISA protocols for beginners' research, two pivotal metrics define success: sensitivity and dynamic range. Sensitivity, or the lower limit of detection (LLOD), determines the smallest analyte quantity distinguishable from background. Dynamic range, the span from the LLOD to the upper limit of quantification (ULOQ), defines the assay's working capacity. Optimizing both is critical for generating reliable, publication-quality data in drug development and basic research. This guide details technical strategies to achieve this optimization.
Foundational Principles: The Binding Curve
The performance of an immunoassay is governed by the law of mass action and the resultant sigmoidal binding curve. Sensitivity is determined by the steepness of the curve's lower asymptote, while dynamic range is defined by the concentration span between its lower and upper plateaus. The core challenge is to shift this curve leftward (increased sensitivity) and expand its linear segment (wider dynamic range).
Strategies for Enhancing Sensitivity
Signal Amplification Systems
Amplifying the detectable signal per binding event is the most direct path to lower detection limits.
Protocol: Tyramide Signal Amplification (TSA) for ELISA
- Materials: Standard ELISA components, hydrogen peroxide, biotinyl-tyramide or fluorescent-tyramide, streptavidin-HRP (if using biotinyl-tyramide).
- Method:
- Perform a standard capture and detection antibody incubation.
- Incubate with HRP-conjugated secondary antibody (1:5000-1:20000 dilution in assay buffer) for 1 hour at RT.
- Wash plates 5 times with PBS-T.
- Prepare tyramide working solution (1-50 µg/mL in amplification diluent containing H₂O₂).
- Incubate 50-100 µL/well of tyramide solution for 2-10 minutes.
- Wash plates thoroughly 5 times with PBS-T.
- If using biotinyl-tyramide: Incubate with streptavidin-HRP (1:1000 dilution) for 30 minutes, wash, then proceed to chromogenic/chemiluminescent detection.
High-Affinity & Mono-Valent Binders
Improving the binding kinetics of the primary antibody directly improves sensitivity by capturing more analyte at low concentrations.
Protocol: Phage Display Selection for High-Affinity ScFv Fragments
- Materials: Phage display library, immobilized target antigen, ELISA plates, blocking buffer, elution buffer, E. coli for infection.
- Method:
- Coat plate with 100 µL/well of purified target antigen (1-10 µg/mL).
- Block with 200 µL/well of 2% BSA/PBS.
- Incubate with 100 µL of phage library for 2 hours.
- Wash 10 times with PBS-T to remove non-binding phage.
- Elute bound phage using 100 µL of 0.1 M glycine-HCl (pH 2.2) for 10 minutes, then neutralize with 15 µL of 1 M Tris-HCl (pH 9.1).
- Amplify eluted phage by infecting log-phase E. coli.
- Repeat panning for 3-5 rounds under increasing stringency (e.g., reduced antigen coating concentration, more washes, competitive elution).
- Screen individual clones by phage ELISA for antigen binding.
Background Noise Reduction
Lowering non-specific signal (noise) improves the signal-to-noise ratio, effectively increasing sensitivity.
Protocol: Optimized Blocking and Wash Stringency
- Method:
- Blocking Agent Screening: Test various blocking agents (e.g., 1% BSA, 5% non-fat dry milk, 1% Casein, commercial protein-free blockers) by running a zero-analyte (blank) control. Select the agent yielding the lowest background OD.
- Wash Optimization: Increase wash volume to 300-350 µL/well. Implement "soak" steps (letting wash buffer sit for 30 seconds) before aspiration. Increase wash cycles from 3 to 5-6 times post critical steps (blocking, sample incubation, detection antibody).
Strategies for Expanding Dynamic Range
Antibody Pair Epitope Bin Analysis
Using a matched pair of antibodies that bind to non-overlapping epitopes ensures the capture and detection of only intact, fully accessible analyte, preventing the "hook effect" and extending the ULOQ.
Protocol: Competitive Epitope Binning via ELISA
- Materials: Biotinylated and non-biotinylated versions of both capture and detection antibodies, streptavidin-HRP.
- Method:
- Coat plate with antigen.
- Pre-mix a constant concentration of biotinylated Antibody A with serial dilutions of non-biotinylated Antibody B (and vice versa). Include self-competition controls.
- Add mixtures to antigen-coated wells and incubate.
- Detect bound biotinylated antibody with streptavidin-HRP.
- If Antibody B competes with Antibody A for binding, signal decreases, indicating epitope overlap. Non-competing pairs are ideal for sandwich ELISA.
Readout Method Selection
The detection chemistry imposes physical limits on the maximum measurable signal.
Table 1: Comparison of ELISA Detection Modalities
| Detection Method | Typical LLOD | Typical Dynamic Range | Key Principle | Best For |
|---|---|---|---|---|
| Chromogenic (TMB) | Moderate | ~2 logs | Enzymatic conversion of substrate to colored soluble product. | Visual assessment, standard plate readers. |
| Chemiluminescent | Very Low | ~3-4+ logs | Enzymatic generation of light photons. | Max sensitivity, widest dynamic range. |
| Electrochemiluminescent (ECL) | Very Low | ~5-6 logs | Ruthenium label excited at electrode surface emits light. | Ultra-high performance assays (e.g., MSD platform). |
| Fluorescent | Low to Moderate | ~3-4 logs | Direct excitation/emission of fluorescent label. | Multiplexing, avoiding enzyme complications. |
Protocol: Transition from Chromogenic to Chemiluminescent Detection
- Materials: Chemiluminescent substrate (e.g., luminol/enhancer/H₂O₂), compatible HRP or AP conjugate, plate reader capable of measuring luminescence.
- Method:
- Perform all ELISA steps up to and including incubation with enzyme-conjugated detection antibody.
- Wash plate as standard.
- Prepare chemiluminescent substrate according to manufacturer's instructions, protecting from light.
- Add substrate to wells (e.g., 100 µL/well). Incubate for 2-5 minutes at RT in the dark.
- Read plate immediately using luminescence settings (integration time 100-500 ms/well).
Data Transformation and Curve Fitting
Mathematical processing of raw data can extract a wider linear range.
Protocol: Four-Parameter Logistic (4PL) Curve Fitting
- Method:
- Collect absorbance/luminescence values for a standard curve with 8-10 points spanning the expected range.
- Using analysis software (GraphPad Prism, SoftMax Pro), fit the data to a 4PL model:
y = d + (a - d) / (1 + (x/c)^b)a= lower asymptoteb= slope factor (Hill coefficient)c= inflection point (EC50)d= upper asymptote
- The linear dynamic range is typically defined as the concentrations corresponding to 10-90% of the response between the asymptotes (ED10 to ED90).
Integrated Experimental Workflow
Workflow for ELISA Optimization
Tyramide Signal Amplification (TSA) Pathway
TSA Mechanism for Signal Amplification
The Scientist's Toolkit: Key Reagent Solutions
| Item | Function & Role in Optimization |
|---|---|
| High-Affinity Matched Antibody Pair | Non-competing capture/detection antibodies specific for distinct epitopes maximize specificity, sensitivity, and dynamic range by ensuring efficient sandwich formation. |
| Chemiluminescent Substrate | An enzymatic substrate (e.g., for HRP or AP) that produces light, offering a wider dynamic range and lower background than chromogenic substrates. |
| Tyramide Amplification Reagent | An enzyme-activated compound that deposits numerous labels (biotin, fluorophore) at the site of antibody binding, dramatically amplifying signal. |
| Protein-Free Blocking Buffer | Reduces non-specific binding from serum components or cross-reactivity, lowering background noise more effectively than protein-based blockers in some assays. |
| Precision Microplate Washer | Ensures consistent and stringent wash cycles, critical for removing unbound material and reducing background variability. |
| Electrochemiluminescence (ECL) Plate | Plates with integrated electrodes (e.g., from MSD) that enable ECL detection, providing the broadest dynamic range and multiplexing capability. |
| 4-Parameter Curve Fitting Software | Essential for accurately modeling the sigmoidal standard curve, determining the linear range, and interpolating unknown sample concentrations. |
Systematic improvement of ELISA sensitivity and dynamic range is achieved through a multi-faceted approach: selecting and engineering high-quality binders, employing advanced signal amplification, rigorously minimizing background, and choosing an appropriate detection system paired with robust data analysis. For the beginner researcher, methodically iterating through these elements transforms a basic protocol into a powerful quantitative tool capable of supporting rigorous scientific inquiry and drug development.
Best Practices for Sample Preparation and Storage to Prevent Degradation
Within the context of developing a robust ELISA protocol for beginners, the integrity of the assay is fundamentally dependent on the quality of the input sample. Improper sample preparation and storage are primary sources of pre-analytical variability, leading to analyte degradation, loss of epitope recognition, and ultimately, unreliable data. This guide details critical, evidence-based practices to preserve sample integrity from collection to analysis.
Core Principles of Sample Integrity
Degradation is driven by enzymatic activity, oxidation, microbial growth, and physical instability. The core tenets to combat these are:
- Inhibition of Proteolysis: Use appropriate protease and phosphatase inhibitors immediately upon collection.
- Temperature Control: Rapid cooling and consistent storage at optimal temperatures.
- Minimization of Freeze-Thaw Cycles: Aliquot samples to avoid repeated cycling.
- Use of Stabilizing Additives: Employ buffers and reagents that maintain pH and molecular conformation.
Quantitative Stability Data by Sample Type
The following table summarizes key stability findings for common sample matrices used in ELISA, informing storage protocols.
Table 1: Stability of Common ELISA Samples Under Various Conditions
| Sample Matrix | Key Analyte(s) | Short-Term (1-7 days) | Long-Term (>1 month) | Critical Inhibitors Needed | Max Recommended Freeze-Thaw Cycles |
|---|---|---|---|---|---|
| Serum | Cytokines, Hormones | 4-8°C | -80°C | Protease inhibitors | 2-3 |
| Plasma (EDTA) | Phosphoproteins, VEGF | 4°C | -80°C | Protease & phosphatase cocktails | 1-2 |
| Cell Lysates | Signaling Proteins | 4°C (with inhibitors) | -80°C | Protease, phosphatase, ubiquitinase inhibitors | 1 |
| Tissue Homogenates | Diverse Proteins | Process immediately | -80°C | Broad-spectrum protease inhibitors | Avoid |
| Cell Culture Supernatant | Secreted Factors | 4°C | -80°C | Azide (if no live cells) | 2-3 |
Detailed Experimental Protocols for Sample Handling
Protocol 3.1: Preparation of Stabilized Plasma for Phospho-protein Analysis
Objective: To collect plasma suitable for phosphorylated epitope detection (e.g., p-STAT, p-ERK) in sandwich ELISA. Materials: EDTA or citrate tubes, pre-chilled centrifuge, protease inhibitor cocktail (PIC), phosphatase inhibitor cocktail (PhosSTOP or equivalent), cryovials.
- Collection: Draw blood into pre-chilled anticoagulant tube. Invert gently.
- Immediate Processing: Centrifuge at 2,000-3,000 x g for 15 minutes at 4°C within 30 minutes of draw.
- Inhibition: Carefully aspirate the plasma layer. Add 1x PIC and 1x phosphatase inhibitor as per manufacturer instructions.
- Aliquoting: Aliquot into pre-labeled cryovials. Use volumes appropriate for a single ELISA run.
- Flash-Freeze: Snap-freeze aliquots in a dry-ice/ethanol bath or liquid nitrogen.
- Storage: Transfer to a -80°C freezer. Record date and freeze-thaw cycle log.
Protocol 3.2: Preparation of Total Protein Cell Lysates for ELISA
Objective: To generate a representative, undegraded total protein lysate from adherent cells. Materials: Cold PBS, RIPA Lysis Buffer, PIC, PhosSTOP, microcentrifuge, sonicator (or needle).
- Wash: Place culture dish on ice. Aspirate media and wash cells twice with ice-cold PBS.
- Lysis: Add cold RIPA buffer supplemented with 1x PIC and 1x PhosSTOP directly to the plate (e.g., 150 µL for a 6-well).
- Harvest: Scrape cells vigorously and transfer the suspension to a pre-chilled microcentrifuge tube.
- Homogenize: Sonicate on ice (3 pulses of 5 seconds) or pass through a 25-gauge needle 10 times.
- Clarify: Centrifuge at 16,000 x g for 15 minutes at 4°C.
- Collect & Aliquot: Transfer the clear supernatant to a new tube. Aliquot immediately.
- Storage: Snap-freeze and store at -80°C. Avoid repeated thawing.
Visualization of Workflows and Concepts
Diagram 1: Sample Integrity Workflow from Collection to ELISA
Diagram 2: Degradation Pathways and ELISA Impact
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Sample Preservation
| Item | Function & Rationale | Key Consideration for ELISA |
|---|---|---|
| Protease Inhibitor Cocktail (PIC) | Broad-spectrum inhibition of serine, cysteine, metallo-proteases. Prevents analyte digestion. | Use compatible, non-interfering formulations. Avoid EDTA in PIC if measuring calcium-dependent analytes. |
| Phosphatase Inhibitor Cocktail | Inhibits serine/threonine and tyrosine phosphatases. Critical for phospho-protein stability. | Essential for any signaling protein assay (e.g., p-kinases). Add fresh to lysis buffer. |
| PMSF (Phenylmethylsulfonyl fluoride) | Serine protease inhibitor. Common, inexpensive addition for basic protein stabilization. | Unstable in aqueous solution; add to buffer just before use. Toxic. |
| EDTA or Citrate Blood Tubes | Chelates calcium to prevent coagulation and inhibit metalloproteases. Preferred for plasma. | EDTA can interfere with some assays (e.g., calcium assays); choose citrate as an alternative. |
| Protease-Free BSA or Azide | Stabilizes dilute protein solutions and prevents microbial growth in supernatants or buffers. | Sodium azide inhibits HRP; do not use in samples for HRP-based ELISA systems. |
| Cryogenic Vials (Screw-cap) | Secure, leak-proof storage for low-temperature preservation. | Use externally threaded caps. Always label with solvent-resistant ink or labels. |
| RIPA Lysis Buffer | Efficiently lyses cells and denatures some proteases while maintaining protein solubility. | May disrupt some protein-protein interactions. For native ELISAs, use milder NP-40 based buffers. |
Validating Your ELISA Results: Standards, Controls, and Data Analysis
Within the foundational thesis on ELISA protocol for beginners, mastering the generation and analysis of the standard curve is paramount. This in-depth technical guide details the construction of a dilution series and the critical evaluation of curve-fitting models—linear and nonlinear (4- and 5-Parameter Logistic, 4/5-PL). A robust standard curve is the cornerstone of accurate quantification in immunoassays, directly impacting data reliability in research and drug development.
Part 1: Designing and Executing the Dilution Series
A serial dilution systematically reduces the concentration of a known standard to create reference points for the assay.
Key Research Reagent Solutions
| Item | Function |
|---|---|
| Reference Standard | Purified analyte of known concentration and high purity. Serves as the calibration anchor. |
| Diluent Matrix | Buffer, often assay-specific, used to dilute the standard. Should mimic the sample matrix to minimize matrix effects. |
| Microplate | 96-well plate, typically clear for colorimetric ELISA, where serial dilutions and samples are dispensed. |
| Multichannel Pipette | Enables rapid, precise, and reproducible transfer of liquid across multiple wells simultaneously. |
| Plate Reader | Instrument that measures the optical density (OD) or other signal (e.g., fluorescence) from each well. |
Detailed Protocol: Two-Fold Serial Dilution
Objective: Prepare a 7-point standard curve from a top concentration of 1000 pg/mL, with a 1:2 dilution factor.
- Preparation: Reconstitute the lyophilized standard as per the datasheet. Prepare a working dilution to achieve the Top Standard Concentration (C1) of 1000 pg/mL in the recommended diluent.
- Plate Layout: Label a microplate for standards (S1-S7) and a blank (diluent only). Each concentration should be assayed in duplicate or triplicate.
- Dilution: Add a volume of diluent (e.g., 100 µL) to wells S2 through S7.
- Transfer: Add an equal volume (e.g., 100 µL) of the 1000 pg/mL top standard (C1) to well S1 and S2.
- Serial Mixing: Mix the contents of well S2 thoroughly. Then, transfer 100 µL from well S2 to well S3. Mix and repeat the process serially through well S7. After mixing well S7, discard 100 µL.
- Blank: The blank well contains diluent only.
- Assay: Proceed with the ELISA protocol (coating, blocking, detection, etc.) for the entire plate.
Data Presentation: Theoretical Dilution Series
Table 1: Example Two-Fold Serial Dilution Series
| Well | Dilution Factor | Relative Concentration | Absolute Concentration (pg/mL) |
|---|---|---|---|
| S1 | 1:1 (Neat) | 1 | 1000 |
| S2 | 1:2 | 1/2 | 500 |
| S3 | 1:4 | 1/4 | 250 |
| S4 | 1:8 | 1/8 | 125 |
| S5 | 1:16 | 1/16 | 62.5 |
| S6 | 1:32 | 1/32 | 31.25 |
| S7 | 1:64 | 1/64 | 15.625 |
| Blank | - | 0 | 0 |
Part 2: Curve Fitting Models: Linear vs. 4/5-PL
After measuring the assay signal (e.g., OD450nm), the standard curve is generated by plotting signal versus concentration and fitting a mathematical model.
Linear Regression
- Application: Used only for data that is linear over the working range. Often achieved by log-transforming one or both axes (e.g., Log(OD) vs. Log(Concentration)).
- Model:
y = mx + c, where y is signal, x is concentration (or log concentration), m is slope, c is y-intercept. - Advantage: Simple, easy to implement.
- Disadvantage: Poorly fits the sigmoidal (S-shaped) response typical of ELISA, especially at the upper and lower asymptotes.
4-Parameter Logistic (4PL) and 5-Parameter Logistic (5PL) Models
These are the industry standard for immunoassay data, accurately describing the sigmoidal dose-response curve.
- 4PL Model:
y = D + (A - D) / (1 + (x/C)^B)- A: Minimum asymptote (background signal).
- B: Hill slope (steepness of the curve).
- C: Inflection point (EC50/IC50).
- D: Maximum asymptote (maximum signal).
- 5PL Model:
y = D + (A - D) / (1 + (x/C)^B)^G- Adds an asymmetry parameter (G), allowing the curve to be asymmetric. Provides superior fit for highly asymmetric data.
Table 2: Comparison of Curve Fitting Models
| Feature | Linear (Log-Log) | 4-Parameter Logistic (4PL) | 5-Parameter Logistic (5PL) |
|---|---|---|---|
| Best For | Data with a linear range | Standard sigmoidal ELISA data | Asymmetric sigmoidal data (common with modern, sensitive assays) |
| Parameters | 2 (Slope, Intercept) | 4 (Min, Max, EC50, Slope) | 5 (Adds Asymmetry) |
| Fit to Plateaus | Poor | Excellent | Superior |
| Complexity | Low | Moderate | High |
| Key Software | Excel, Prism, SoftMax Pro | Prism, SoftMax Pro, PLA | PLA, some versions of Prism & SoftMax Pro |
Detailed Protocol: Curve Fitting and Sample Interpolation
- Data Entry: Input the mean OD for each standard concentration into analysis software (e.g., GraphPad Prism, BioTek Gen5, SoftMax Pro).
- Model Selection: Choose the fitting model (4PL is the default recommendation for beginners). Select appropriate weighting (e.g., 1/Y²) if replicates show non-constant variance.
- Fit the Curve: Execute the regression. Visually inspect that the curve passes near all data points, especially at the lower limit of quantification (LLOQ) and upper limit of quantification (ULOQ). Evaluate the R² or sum of squares.
- Quality Criteria: The curve should have an R² > 0.99. The back-calculated concentrations of standards should be within 20% of expected (15% for LLOQ/ULOQ).
- Interpolate Unknowns: For unknown samples, the software uses the fitted model's equation to calculate the concentration from the measured OD.
Visualizing the ELISA Workflow and Curve Fitting Logic
Diagram 1: ELISA Standard Curve Generation and Analysis Workflow
Diagram 2: Curve Fitting Model Comparison and Selection
Within a beginner's ELISA research thesis, the establishment of a robust, validated protocol is paramount. Central to this validation is the strategic implementation of specific controls. These controls are not mere procedural steps; they are the diagnostic tools that verify every component of the assay system, from reagent functionality to procedural accuracy. This guide details the core controls—Positive, Negative, Blank, and Spike-in Recovery—framing them as essential elements for generating credible, interpretable data in quantitative immunoassays.
Core Control Definitions and Functions
- Positive Control: A sample with a known, detectable concentration of the target analyte. It confirms that the assay procedure and all reagents (antibodies, enzyme conjugates, substrates) are functional. A failure of the positive control invalidates the entire experimental run.
- Negative Control: A sample confirmed to lack the target analyte. It establishes the baseline signal in the absence of specific binding and is critical for assessing non-specific background interference.
- Blank Control: A well where all assay steps are performed except the addition of any sample or control. Typically, only buffer is added. It measures the background signal inherent to the assay reagents and substrate.
- Spike-in Recovery Control: A sample of known, negative matrix (e.g., serum, cell lysate) into which a known quantity of the target analyte has been added ("spiked"). It is used to assess the accuracy of the assay in the presence of the sample's complex background, detecting matrix interference effects.
Table 1: Expected Outcomes and Interpretation of Core ELISA Controls
| Control Type | Purpose | Expected Result (vs. Standard Curve) | Typical Acceptance Criterion | Outcome Interpretation |
|---|---|---|---|---|
| Positive Control | Verify assay functionality | Quantifiable concentration within expected range. | Recovery within 80-120% of stated value. | Fail: Assay reagents or steps compromised. Results invalid. |
| Negative Control | Assess non-specific binding | Concentration below the Lower Limit of Quantification (LLOQ). | Signal ≤ LLOQ or ≤ 20% of positive control. | High Signal: Indicates antibody cross-reactivity or matrix interference. |
| Blank (Reagent Blank) | Define reagent background | Zero concentration. Optical Density (OD) near substrate blank. | OD ≤ 10% of mean positive control OD. | High OD: Contamination or non-specific substrate activation. |
| Spike-in Recovery | Measure matrix interference | Measured concentration of spike matches expected value. | Recovery of 70-130% (matrix-dependent). | Low Recovery: Matrix components inhibit detection. High Recovery: Non-specific signal enhancement. |
Table 2: Example Spike-in Recovery Experiment Data
| Sample Matrix | Endogenous [Analyte] | Spike Amount Added | Total Expected [Analyte] | Measured [Analyte] | % Recovery | Assessment |
|---|---|---|---|---|---|---|
| Calibration Diluent | 0 ng/mL | 50 ng/mL | 50 ng/mL | 49.2 ng/mL | 98.4% | Ideal (No matrix) |
| Normal Serum 1 | 50 ng/mL | 50 ng/mL | 42.5 ng/mL | 85.0% | Acceptable | |
| Normal Serum 2 | 50 ng/mL | 50 ng/mL | 32.0 ng/mL | 64.0% | Unacceptable - Matrix Inhibition | |
| Cell Lysis Buffer | 0 ng/mL | 50 ng/mL | 50 ng/mL | 61.0 ng/mL | 122.0% | Unacceptable - Signal Enhancement |
Detailed Experimental Protocols
Protocol 1: Implementing Standard Controls in a Sandwich ELISA
- Plate Layout: Designate wells on the microplate for Blank (B), Negative Control (NC), Positive Control (PC), and samples in duplicate or triplicate.
- Coating: Coat plate with capture antibody. Wash.
- Blocking: Add blocking buffer (e.g., 5% BSA/PBS) to all wells, including future Blank well. Incubate. Wash.
- Sample Addition:
- Blank (B): Add sample diluent/buffer only.
- Negative Control (NC): Add matrix confirmed to be analyte-negative.
- Positive Control (PC): Add provided control or spiked matrix.
- Samples: Add test samples. Incubate. Wash.
- Detection Antibody Addition: Add enzyme-conjugated detection antibody to all wells except the designated Blank (B) well. Add buffer to B well. Incubate. Wash.
- Substrate Addition: Add enzyme substrate (e.g., TMB) to ALL wells, including Blank. Incubate in dark.
- Stop & Read: Add stop solution. Read Optical Density (OD) immediately.
- Analysis: Generate standard curve. Calculate concentrations for PC and samples. NC and B should fall below the LLOQ.
Protocol 2: Conducting a Spike-in Recovery Experiment
- Preparation of Spike Solution: Prepare a concentrated solution of the pure analyte in an appropriate buffer.
- Selection of Baseline Matrix: Obtain multiple lots of the intended sample matrix (e.g., serum from ≥3 donors) known to be negative or low for the analyte.
- Spiking:
- Prepare a "Matrix Spike" sample: Add a precise volume of spike solution to the negative matrix. The final concentration should be within the assay's quantitative range (e.g., mid-point of standard curve).
- Prepare a "Reference Spike" sample: Add the same volume of spike solution to the assay's calibrator diluent (a non-interfering buffer).
- Prepare an "Unspiked Matrix" sample: Add buffer only to the same negative matrix.
- Assay Execution: Run all samples (Spiked Matrix, Reference Spike, Unspiked Matrix) in the same ELISA plate alongside the standard curve, in replicate (n≥3).
- Calculation:
- Calculate the measured concentration for each sample from the standard curve.
- % Recovery = ( [Spiked Matrix] - [Unspiked Matrix] ) / [Reference Spike] × 100
Visualizations
Title: ELISA Control Validation Logic Flow
Title: Spike-in Recovery Experimental Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Control Implementation in ELISA
| Reagent / Material | Function in Control Context | Critical Specification |
|---|---|---|
| Analyte-Negative Matrix | Serves as the base for Negative Control and for preparing Spike-in samples. Must be validated as truly negative. | Pooled from multiple donors/lots to ensure representativeness. |
| Recombinant Purified Analyte | Used to prepare Positive Control and Spike-in solutions. | High purity (>95%) and verified biological activity. |
| Assay Diluent / Buffer | Used for Blank control, sample dilution, and preparing the Reference Spike. | Optimized to minimize non-specific binding; protein-based (e.g., BSA). |
| Validated Positive Control | Commercially available or internally prepared control of known concentration. | Concentration should be near the midpoint of the standard curve. Stability documented. |
| Monoclonal Antibody Pair | Capture and detection antibodies form the basis of sandwich ELISA specificity. | High affinity and specificity; minimal cross-reactivity. |
| Enzyme-Substrate System (e.g., HRP/TMB) | Generates the measurable colorimetric signal. | Low background, high signal-to-noise ratio. Consistent lot-to-lot. |
| Microplate Washer & Reader | For consistent washing and accurate optical density measurement. | Precision and accuracy validated. Proper maintenance is crucial. |
Calculating Sample Concentrations and Assessing Assay Precision (CV%)
For researchers embarking on ELISA-based projects, particularly beginners, mastering two core analytical skills is paramount: accurately calculating the concentration of an analyte in a sample and rigorously assessing the precision of the assay itself. This guide, framed within a foundational thesis on ELISA protocols, details the methodologies for these critical tasks. Accurate concentration determination is the goal of the assay, while precision measurement via the coefficient of variation (CV%) validates the reliability of the obtained data, forming the bedrock of credible research and drug development.
Core Concepts: The Standard Curve and CV%
The standard curve is the linchpin for converting raw optical density (OD) readings into meaningful concentration values. It is generated by assaying samples of known concentration (standards). The relationship between OD and concentration is typically modeled using a 4- or 5-parameter logistic (4PL/5PL) curve fit, which accounts for the non-linear sigmoidal response of ELISA.
Precision is assessed by calculating the Coefficient of Variation (CV%), which expresses the standard deviation as a percentage of the mean. It is the key metric for evaluating assay repeatability (intra-assay precision) and reproducibility (inter-assay precision). A lower CV% indicates higher precision.
Experimental Protocols
Protocol for Generating a Standard Curve
- Reconstitution and Serial Dilution: Precisely reconstitute the standard protein according to the kit datasheet in the recommended matrix (e.g., assay diluent). Perform a serial dilution (e.g., two-fold or five-fold) to create a minimum of 5-7 non-zero standard points covering the entire dynamic range of the assay.
- Plate Layout: Include the standard points, blank (matrix only), and quality controls (QCs) in duplicate or triplicate across the plate.
- Assay Execution: Run the complete ELISA protocol (coating, blocking, sample/standard incubation, detection, substrate development) as per established procedures.
- Data Acquisition: Measure the OD for each well using a plate reader at the appropriate wavelength.
Protocol for Assessing Intra- and Inter-Assay Precision
- Sample Preparation: Prepare three QC samples (low, mid, and high concentration) within the assay's range using the analyte spiked into the appropriate matrix.
- Intra-Assay Precision: In a single assay run, analyze each QC sample in a minimum of 6-8 replicates. Calculate the mean, standard deviation (SD), and CV% for each QC level.
- Inter-Assay Precision: Across multiple independent assay runs (at least 3 different days, with different operators or reagent lots if possible), analyze the same QC samples in duplicate or triplicate per run. Pool all results for each QC level and calculate the overall mean, SD, and CV%.
Data Analysis & Presentation
Calculating Sample Concentration
- Curve Fitting: Input the mean OD values for each standard into analysis software (e.g., GraphPad Prism, SoftMax Pro). Generate a 4PL or 5PL curve fit.
- Interpolation: For each unknown sample, use its mean OD value to interpolate the concentration from the standard curve equation. Most software performs this automatically.
- Dilution Factor: Multiply the interpolated concentration by any sample dilution factor applied prior to the assay.
Assessing Precision: CV% Calculation
The CV% is calculated for each QC level (and for replicate standards to assess curve precision) using the formula: CV% = (Standard Deviation / Mean) x 100
Table 1: Example Intra-Assay Precision Data for a Cytokine ELISA
| QC Level | Nominal Conc. (pg/mL) | Replicate Measurements (pg/mL) | Mean (pg/mL) | SD (pg/mL) | CV% |
|---|---|---|---|---|---|
| Low | 50 | 48, 52, 49, 51, 47, 53 | 50.0 | 2.2 | 4.4 |
| Mid | 200 | 195, 208, 202, 198, 205, 192 | 200.0 | 5.7 | 2.9 |
| High | 800 | 790, 810, 805, 788, 812, 795 | 800.0 | 9.8 | 1.2 |
Table 2: Example Inter-Assay Precision Summary
| QC Level | Overall Mean (pg/mL) | Overall SD (pg/mL) | CV% | Number of Runs |
|---|---|---|---|---|
| Low | 49.5 | 3.5 | 7.1 | 5 |
| Mid | 198 | 8.2 | 4.1 | 5 |
| High | 805 | 15.3 | 1.9 | 5 |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Quantitative ELISA Analysis
| Item | Function |
|---|---|
| Reference Standard | A purified analyte of known concentration and activity, essential for constructing the standard curve. |
| Matrix-Matched Diluent | A buffer or solution that mimics the sample matrix (e.g., serum, cell lysate) to minimize matrix effects in standard and sample dilution. |
| Quality Control (QC) Samples | Pre-prepared samples with known analyte concentrations used to verify assay performance and precision across runs. |
| Precision Pipettes & Tips | For accurate and reproducible liquid handling during serial dilution and sample/reagent transfer. |
| Microplate Reader | Instrument to measure the optical density (absorbance) of each well, generating the raw data for analysis. |
| Curve-Fitting Software | Specialized software (e.g., ELISA analysis modules) that applies appropriate regression models (4PL/5PL) to the standard data for accurate sample interpolation. |
Visualizing Workflows and Relationships
ELISA Data Analysis & Precision Workflow
CV% Calculation Logic
Within a foundational thesis on ELISA protocol for beginners, it is critical to understand that the enzyme-linked immunosorbent assay (ELISA) is often the first immunoassay mastered. Its principles of antigen-antibody binding and enzymatic detection form the bedrock of immunodetection. However, as research questions evolve in complexity—requiring multiplexing, superior sensitivity, or protein size information—alternatives like Western Blot, Luminex, and Mesoscale Discovery (MSD) assays become necessary. This guide provides an in-depth technical comparison to inform assay selection.
Core Assay Comparison
Table 1: High-Level Technical Comparison of Immunoassays
| Feature | ELISA | Western Blot | Luminex/xMAP | MSD/ECLIA |
|---|---|---|---|---|
| Primary Output | Quantification of soluble analyte | Detection & size estimation of proteins | Multiplex quantification of soluble analytes | Quantification of soluble analytes |
| Throughput | High (96/384-well) | Low to Medium | Very High (up to 500-plex) | High (96/384-well) |
| Multiplex Capability | Singleplex (typically) | Semi-multiplex (limited by band proximity) | High (10-500 targets) | Medium (typically up to 10-plex) |
| Sensitivity | Moderate (pg/mL) | Low to Moderate (ng/mL) | Moderate to High (pg/mL) | Very High (fg/mL - pg/mL) |
| Dynamic Range | ~2-3 logs | ~1.5 logs | ~3-4 logs | >4 logs |
| Sample Type | Serum, plasma, supernatant, lysate* | Cell/tissue lysate (denatured) | Serum, plasma, supernatant, lysate* | Serum, plasma, supernatant, lysate* |
| Key Advantage | Simple, standardized, cost-effective | Confirms molecular weight, detects isoforms | High-level multiplexing in small sample volume | Excellent sensitivity & dynamic range, low background |
| Key Limitation | Single analyte, potential cross-reactivity | Low throughput, qualitative/semi-quantitative | Complex data analysis, reagent availability | Higher cost per sample, specialized equipment |
*Requires specific lysate buffers compatible with the assay format.
Detailed Methodologies and Applications
ELISA (Enzyme-Linked Immunosorbent Assay)
Protocol (Direct Sandwich ELISA):
- Coating: Dilute capture antibody in carbonate/bicarbonate coating buffer (pH 9.6). Add 100 µL/well to a 96-well microplate. Seal and incubate overnight at 4°C.
- Washing: Aspirate and wash plate 3x with 300 µL/well of PBS containing 0.05% Tween-20 (PBST).
- Blocking: Add 200 µL/well of blocking buffer (e.g., 5% BSA or non-fat dry milk in PBS). Incubate 1-2 hours at room temperature (RT). Wash 3x.
- Sample/Antigen Incubation: Add 100 µL/well of standard dilutions or samples diluted in assay diluent. Incubate 2 hours at RT. Wash 3x.
- Detection Antibody Incubation: Add 100 µL/well of biotin-conjugated detection antibody. Incubate 1-2 hours at RT. Wash 3x.
- Streptavidin-Enzyme Conjugate: Add 100 µL/well of Streptavidin-Horseradish Peroxidase (HRP) diluted in assay diluent. Incubate 30 minutes at RT. Wash 3x.
- Substrate Addition: Add 100 µL/well of TMB substrate. Incubate 5-20 minutes at RT in the dark.
- Stop and Read: Add 50 µL/well of stop solution (e.g., 2N H₂SO₄). Read absorbance immediately at 450 nm with a correction wavelength of 570 or 620 nm.
Western Blot (Immunoblot)
Protocol:
- Sample Preparation: Lyse cells/tissue in RIPA buffer with protease inhibitors. Determine protein concentration via BCA assay. Mix 20-50 µg protein with Laemmli buffer, denature at 95°C for 5 min.
- Gel Electrophoresis: Load samples onto a polyacrylamide gel (SDS-PAGE). Run at constant voltage (e.g., 80-120V) until dye front reaches bottom.
- Protein Transfer: Transfer proteins from gel to PVDF or nitrocellulose membrane using wet or semi-dry transfer apparatus.
- Blocking: Incubate membrane in 5% non-fat milk in TBST for 1 hour at RT.
- Primary Antibody Incubation: Incubate membrane with primary antibody diluted in blocking buffer overnight at 4°C.
- Washing: Wash membrane 3 x 10 min with TBST.
- Secondary Antibody Incubation: Incubate with HRP-conjugated secondary antibody (e.g., anti-rabbit IgG) for 1 hour at RT. Wash 3 x 10 min.
- Detection: Incubate membrane with chemiluminescent substrate (e.g., ECL). Image using a CCD camera-based imager.
Luminex/xMAP Technology
Protocol (Sandwich Immunoassay on Magnetic Beads):
- Bead Coupling: Carboxylated magnetic beads are chemically coupled to capture antibodies using EDC/sulfo-NHS chemistry.
- Assay Setup: In a 96-well plate, combine 50 µL of sample/standard with 50 µL of antibody-coupled bead mixture. Seal, incubate 2 hours on a plate shaker.
- Washing: Place plate on a magnetic separator. Discard supernatant. Wash beads 2x with wash buffer.
- Detection Antibody Incubation: Add 50 µL of biotinylated detection antibody. Incubate 1 hour on shaker. Wash 2x.
- Streptavidin-Phycoerythrin (SA-PE) Incubation: Add 50 µL of SA-PE. Incubate 30 min on shaker. Wash 2x.
- Resuspension & Reading: Resuspend beads in 100-150 µL of drive fluid. Analyze on a Luminex analyzer, which identifies each bead by its internal fluorescent signature and quantifies the bound analyte via PE fluorescence.
MSD (Mesoscale Discovery) Electrochemiluminescence
Protocol (ULTRA-Sensitive Sandwich Assay):
- Plate Coating: MSD plates contain carbon electrode surfaces. Add 30 µL/well of capture antibody solution. Incubate sealed plate 1 hour at RT with shaking.
- Blocking: Aspirate, add 150 µL/well of MSD Blocker A solution. Incubate 30 min with shaking. Wash 3x with PBST.
- Sample/Antigen Incubation: Add 25 µL/well of standard or sample. Add 25 µL/well of detection antibody conjugated with an electrochemiluminescent label (MSD SULFO-TAG). Incubate 2 hours with shaking. Wash 3x.
- Reading: Add 150 µL/well of MSD GOLD Read Buffer B. The plate is immediately read in an MSD instrument. An electrical voltage is applied, triggering the SULFO-TAG label to emit light, which is measured.
Decision Framework and Visualization
Decision Workflow for Immunoassay Selection
Western Blot Experimental Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Immunoassays
| Item | Primary Function | Key Considerations |
|---|---|---|
| Microplates (ELISA/MSD) | Solid phase for assay immobilization. | Material (polystyrene, high-binding), well format (96/384), MSD plates have integrated electrodes. |
| Nitrocellulose/PVDF Membranes (WB) | Porous substrate for protein immobilization post-transfer. | Pore size (0.2-0.45 µm), PVDF requires methanol activation, offers better protein retention. |
| Magnetic Beads (Luminex) | Solid support with spectral barcode for multiplexing. | Bead region (analyte identity), surface chemistry (carboxyl, streptavidin), magnetic core for washing. |
| Capture & Detection Antibodies | Specific antigen recognition. | Matched pair (non-competing epitopes) for sandwich assays; species/host and clonality critical. |
| Enzyme Conjugates | Signal generation. | HRP or Alkaline Phosphatase (AP) linked to antibody or streptavidin. Stability varies. |
| Electrochemiluminescent Label (MSD SULFO-TAG) | Ruthenium-based label emits light upon electrical stimulation. | Covalently linked to detection antibody. Enables low-background, sensitive detection. |
| Phycoerythrin (PE) Conjugate (Luminex) | Fluorescent reporter molecule. | Streptavidin-PE is common; photostability is a consideration. |
| Chemiluminescent Substrate | Enzyme substrate producing light signal. | Enhanced substrates (e.g., ECL Prime) offer higher sensitivity. Luminol-based for HRP. |
| Blocking Buffers | Prevent non-specific binding. | Protein-based (BSA, casein, serum) or synthetic; must be optimized for each assay. |
| Signal Readout Instrument | Quantifies assay signal. | Plate reader (Abs/FL), CCD imager (WB), Luminex analyzer, MSD SECTOR Imager. |
Within the thesis context of "ELISA Protocol for Beginners Research," establishing data reliability is not an optional step but the very foundation of valid scientific conclusions. For novices, the Enzyme-Linked Immunosorbent Assay (ELISA) presents a powerful yet intricate tool where small procedural variations can lead to significant data misinterpretation. This guide deconstructs the core pillars of reliable ELISA data: Reproducibility (the ability to repeat the experiment and obtain consistent results), Specificity (the assay's ability to measure only the target analyte), and Acceptance Criteria (pre-defined, objective benchmarks for validating a run). Mastery of these concepts transforms a beginner from merely following steps to critically executing and evaluating an assay.
Reproducibility: The Benchmark of Robust Science
Reproducible data instills confidence. In ELISA, reproducibility is achieved through rigorous standardization and documentation.
Key Methodologies for Enhancing Reproducibility:
Precision (Repeatability & Intermediate Precision) Experiments: To quantify assay variability, perform a precision experiment.
- Protocol: Prepare a sample with analyte concentration in the mid-range of the standard curve. Aliquot this sample into multiple wells across the plate (e.g., 8-12 wells). Repeat this entire process on three separate days, with different reagent preparations, by the same analyst (intermediate precision).
- Data Analysis: Calculate the mean concentration, standard deviation (SD), and coefficient of variation (%CV) for each day (repeatability) and for all data pooled together (intermediate precision). Industry standards often target a %CV of <10-15% for precision.
Robust Standard Curve Practices: The standard curve is the anchor of quantification.
- Protocol: Always run a fresh standard curve in duplicate or triplicate on every plate. Use a serial dilution (typically 1:2 or 1:3) covering the expected sample range. Fit the data using appropriate regression (e.g., 4- or 5-parameter logistic). Document the fitted equation and the R² value (goodness-of-fit).
Quantitative Data Summary: Precision Performance Metrics
| Performance Parameter | Calculation | Typical ELISA Acceptance Criterion |
|---|---|---|
| Repeatability (Intra-assay %CV) | (SD of replicates within a plate / Mean) x 100 | ≤ 10-12% |
| Intermediate Precision (Inter-assay %CV) | (SD of all replicates across days / Grand Mean) x 100 | ≤ 15-20% |
| Standard Curve Fit (R²) | Coefficient of determination from regression | ≥ 0.990 |
Specificity: Confirming the Target is Measured
Specificity ensures the signal originates from the intended antigen-antibody interaction. Lack of specificity is a major pitfall for beginners.
Key Methodologies for Demonstrating Specificity:
Spike-and-Recovery and Linearity-of-Dilution: Assesses matrix interference.
- Protocol (Spike/Recovery): "Spike" a known amount of pure analyte into the sample matrix (e.g., serum, cell lysate). Measure the concentration and calculate the percentage recovery:
(Measured Concentration of Spike / Expected Concentration) x 100. Acceptance is typically 80-120%. - Protocol (Linearity-of-Dilution): Serially dilute a sample with high analyte concentration using the appropriate assay buffer or matrix. The measured concentrations, when corrected for dilution, should be constant. Significant trends indicate interference.
- Protocol (Spike/Recovery): "Spike" a known amount of pure analyte into the sample matrix (e.g., serum, cell lysate). Measure the concentration and calculate the percentage recovery:
Cross-Reactivity Testing: Evaluates antibody specificity against structurally similar molecules.
- Protocol: Run the standard curve with the target analyte. In parallel, run curves for potential interfering substances at high concentrations. Calculate the percentage cross-reactivity:
(Concentration of Target at 50% B/B₀ / Concentration of Interferent at 50% B/B₀) x 100. Values <1-5% indicate high specificity.
- Protocol: Run the standard curve with the target analyte. In parallel, run curves for potential interfering substances at high concentrations. Calculate the percentage cross-reactivity:
Defining and Applying Acceptance Criteria
Acceptance criteria are pre-defined, quantitative rules that determine if an ELISA run is valid. They move validation from subjective judgment to objective science.
Core Acceptance Criteria for Each Run:
- Standard Curve Fit: R² ≥ 0.990.
- Back-Calculated Standards: The concentration of each standard, calculated from the curve fit, should be within ±20% of its theoretical value (±25% for the Lower Limit of Quantification).
- Quality Control (QC) Samples: Include at least two levels of QC samples (Low, Mid, High) in duplicate on every plate. ≥67% of QCs (and ≥50% at each level) must fall within their established target ranges (e.g., mean ± 2SD).
Experimental Protocol for Establishing QC Ranges:
- Over at least 5-10 independent assays, measure the prepared QC samples.
- Calculate the mean and SD for each QC level.
- Define the acceptable range as mean ± 2SD or mean ± 3SD, depending on required stringency.
Visualizing the Workflow and Logical Framework
Diagram: ELISA Data Reliability Decision Pathway
Diagram: Key ELISA Validation Parameter Relationships
The Scientist's Toolkit: Research Reagent Solutions
| Essential Material/Reagent | Primary Function in Ensuring Reliability |
|---|---|
| High-Affinity, Validated Antibody Pair | The cornerstone of specificity. Matched capture and detection antibodies minimize cross-reactivity and background. |
| Reference Standard of Known Purity | Enables accurate quantification and calibration of every run. Essential for reproducible standard curves. |
| Matrix-Matched Calibrators/Diluent | Calibrators prepared in the same matrix as samples (e.g., serum) account for interference, improving specificity and recovery. |
| Precision Quality Control (QC) Samples | Pooled samples with low, mid, and high analyte concentrations. The benchmark for acceptance criteria and monitoring long-term precision. |
| Blocking Buffer (e.g., BSA, Casein) | Reduces non-specific binding of antibodies to the plate, lowering background noise and enhancing specificity and signal-to-noise ratio. |
| Plate Washer & Consistent Wash Buffer | Removes unbound reagents uniformly. Critical for reproducibility by minimizing well-to-well variability. |
| Calibrated Multichannel/Microplate Pipettes | Ensures accurate and reproducible liquid handling, especially for serial dilutions and replicate sampling. |
| Validated Substrate (e.g., TMB) & Stop Solution | Provides consistent enzymatic signal generation and termination. Batch-to-batch validation supports reproducible kinetics and optical density readings. |
By systematically implementing these principles of reproducibility, specificity, and predefined acceptance criteria, beginner ELISA researchers can generate data that is not only publishable but fundamentally trustworthy, forming a solid foundation for their broader research thesis.
Conclusion
Mastering the ELISA protocol is a fundamental skill that opens doors to quantitative protein analysis in diverse research and clinical settings. This guide has walked you through the core principles, a robust step-by-step methodology, essential troubleshooting tactics, and rigorous validation practices. By integrating these four pillars—understanding the assay's foundation, executing a meticulous protocol, proactively solving problems, and critically validating results—beginners can transition to competent practitioners. The continued evolution of ELISA, including automation and multiplexing, ensures its enduring relevance. A well-executed ELISA provides not just data, but reliable, interpretable insights that can drive hypothesis testing in basic science, biomarker discovery, and critical quality control in biopharmaceutical development.